Neuroprotective mechanisms of dieckol against glutamate toxicity through reactive oxygen species scavenging and nuclear factor-like 2/heme oxygenase-1 pathway
- Affiliations
-
- 1Department of Physiology, Jeju National University School of Medicine, Jeju 63243, Korea. syeun@jejunu.ac.kr
- 2Department of Neurosurgery, Jeju National University School of Medicine, Jeju 63243, Korea.
- 3Division of Hematology-Oncology, Department of Internal Medicine, Jeju National University School of Medicine, Jeju 63243, Korea.
- 4Neurology 1, The Second Affiliated Hospital of Xinxiang Medical University, Henan 453002, China.
- 5BotaMedi Inc., Jeju 63309, Korea.
- 6Center for Cognition and Sociality, Institute for Basic Science (IBS), KAIST, Daejeon 34126, Korea.
- 7University of Science and Technology, Daejeon 34113, Korea.
- 8Institute of Medical Science, Jeju National University, Jeju 63243, Korea.
- KMID: 2438086
- DOI: http://doi.org/10.4196/kjpp.2019.23.2.121
Abstract
- Glutamate toxicity-mediated mitochondrial dysfunction and neuronal cell death are involved in the pathogenesis of several neurodegenerative diseases as well as acute brain ischemia/stroke. In this study, we investigated the neuroprotective mechanism of dieckol (DEK), one of the phlorotannins isolated from the marine brown alga Ecklonia cava, against glutamate toxicity. Primary cortical neurons (100 µM, 24 h) and HT22 neurons (5 mM, 12 h) were stimulated with glutamate to induce glutamate toxic condition. The results demonstrated that DEK treatment significantly increased cell viability in a dose-dependent manner (1-50 µM) and recovered morphological deterioration in glutamate-stimulated neurons. In addition, DEK strongly attenuated intracellular reactive oxygen species (ROS) levels, mitochondrial overload of Ca²âº and ROS, mitochondrial membrane potential (ΔΨ(m)) disruption, adenine triphosphate depletion. DEK showed free radical scavenging activity in the cell-free system. Furthermore, DEK enhanced protein expression of heme oxygenase-1 (HO-1), an important anti-oxidant enzyme, via the nuclear translocation of nuclear factor-like 2 (Nrf2). Taken together, we conclude that DEK exerts neuroprotective activities against glutamate toxicity through its direct free radical scavenging property and the Nrf-2/HO-1 pathway activation.
Keyword
MeSH Terms
Figure
-
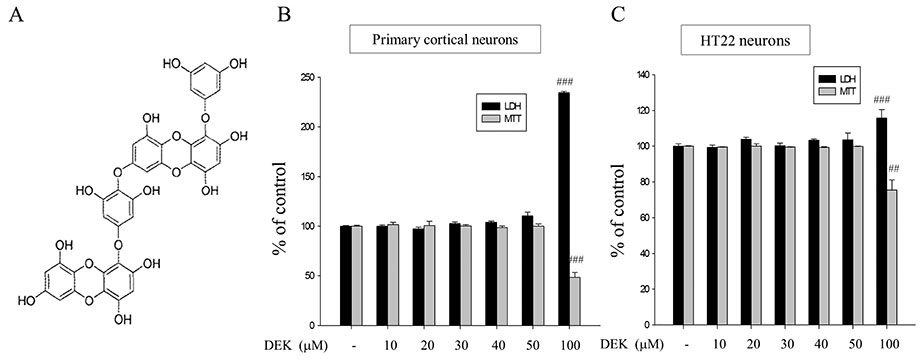
Fig. 1 Chemical structure and cytotoxicity test of DEK. (A) Chemical structure of DEK. (B) Cytotoxicity test of DEK in neurons. Different doses of DEK were treated for 25 h in primary cortical neurons and for 13 h in HT22 neurons. These treatment durations are the sum of co-treatment duration (24 h for primary cortical neurons and 12 h for HT22 cell line) and the DEK pretreatment duration (1 h). The cytotoxicity of DEK was examined using the MTT assay and LDH release assay. DEK below 100 µM did not show any cytotoxicity as a chemical agent for treatment, based on MTT assay and LDH release assay. Values were expressed as mean ± standard error of mean of four samples in one independent experiment. ###p < 0.001, as compared to the untreated control group. DEK, dieckol; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; LDH, lactate dehydrogenase release.

Fig. 2 Neuroprotective effects of DEK on neuronal cell viabilities and morphological changes against glutamate toxicity. (A and B) Both primary cortical neurons and HT22 neuronal cell line were pretreated with DEK for 1 h prior to glutamate stimulation. Then, primary cortical neurons were stimulated with glutamate (100 µM) in the presence of different doses of DEK for 24 h. In case of HT22 neurons, cells were stimulated with glutamate (5 mM) for 12 h. Neuroprotective effects of DEK against glutamate toxicity were evaluated using MTT cell viability assay in primary cortical neurons and HT22 neurons. (C) Representative phase contrast images indicating neuroprotective effects of DEK on glutamate-induced morphological changes in HT22 neurons. Scale bar, 50 µm. Values were expressed as mean ± standard error of mean of four samples in one independent experiment. Statistical analyses were performed using one-way ANOVA followed by Bonferroni test. ###p < 0.001, as compared to untreated control group; *p < 0.05 and ***p < 0.001, as compared to glutamate alone-treated group. DEK, dieckol; Glu, glutamate; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide.
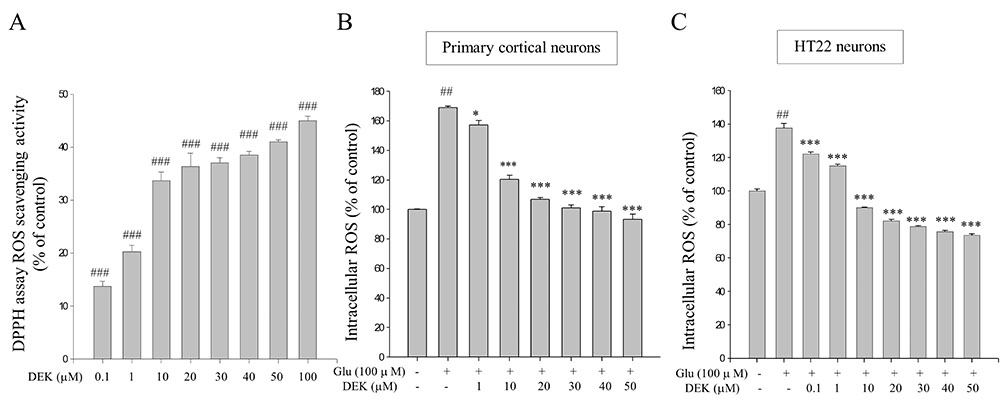
Fig. 3 ROS scavenging activities of DEK in glutamate-stimulated neurons. (A) Free radical scavenging activities of DEK in a cell-free system were performed using the DPPH assay. (B and C) Both primary cortical neurons and HT22 neuronal cell line were pretreated with DEK for 1 h prior to glutamate stimulation. Then, primary cortical neurons were stimulated with glutamate (100 µM) in the presence of different doses of DEK for 24 h. In case of HT22 neurons, cells were stimulated with glutamate (5 mM) for 12 h. Effects of DEK on glutamate-induced intracellular ROS generation were examined using a spectrofluorometer with the ROS-sensitive fluorescent dye DCF-DA. Values were expressed as mean ± standard error of mean of four samples in one independent experiment. Statistical analyses were performed using one-way ANOVA followed by Bonferroni test. ##p < 0.01 and ###p < 0.001, as compared to the untreated control group; *p < 0.05 and ***p < 0.001, as compared to the glutamate alone-treated group. ROS, reactive oxygen species; DPPH, 1,1-diphenyl-2-picrylhydrazyl; DEK, dieckol; Glu, glutamate.
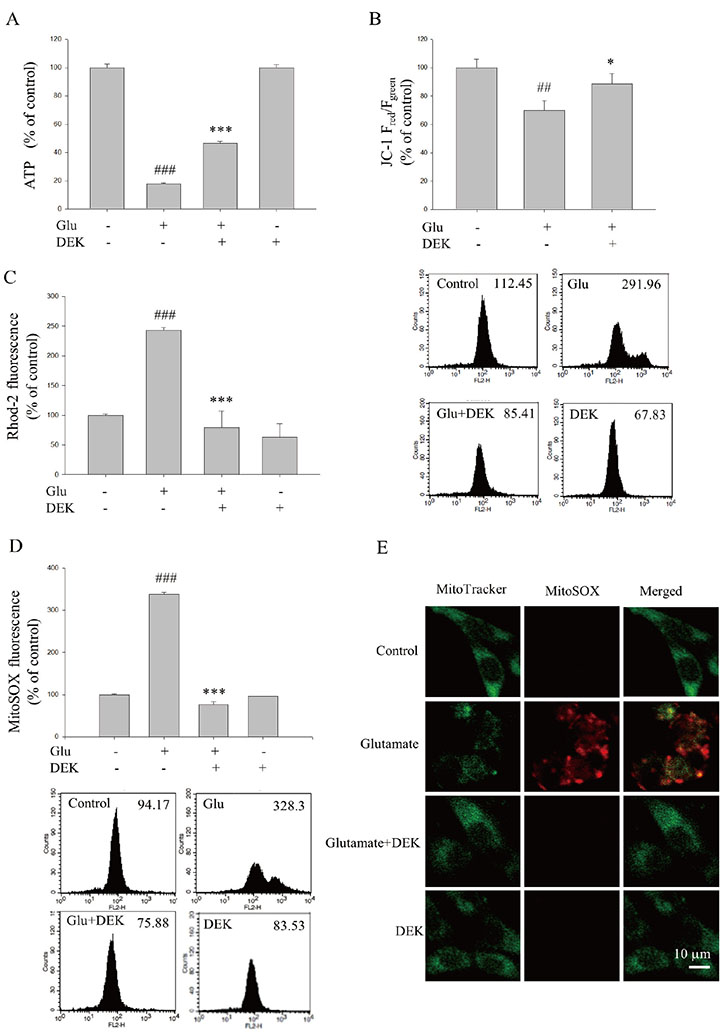
Fig. 4 DEK attenuates glutamate-induced mitochondria dysfunction. HT22 neurons were pretreated with DEK (50 µM) for 1 h and then stimulated with glutamate (5 mM) in the presence of DEK for 12 h. Effects of DEK on glutamate-induced mitochondrial dysfunction were examined. (A) Intracellular ATP levels were measured using a luciferase/luciferin ATP determination kit. (B) The effects of DEK on glutamate-induced ΔΨmm disruption were analyzed by flow cytometry using JC-1. Figure shows the representative data of flow cytometry in the lower panel and their quantitative analyses in the upper panel. (C) The effects of DEK on glutamate-induced mitochondrial Ca2+ levels were analyzed by flow cytometry using Rhod-2. (D) The effects of DEK on glutamate-induced mitochondrial ROS generation were analyzed by flow cytometry using MitoSOX. Figure shows the representative data of flow cytometry in the lower panel and their quantitative analyses in the upper panel. (E) Representative confocal images of HT22 neurons loaded with MitoSOX were shown to demonstrate neuroprotective effects of DEK on glutamate-induced mitochondrial ROS generation. Mitochondria were stained with Mito Tracker Green. Scale bar, 50 µm. Values were expressed as mean ± standard error of mean of four samples in one independent experiment. ##p < 0.01 and ###p < 0.001, as compared to untreated control group; *p < 0.05 and ***p < 0.001, as compared to glutamate alone-treated group. ATP, adenine triphosphate; Glu, glutamate; DEK, dieckol; JC-1, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide; Rhod-2, rhod-2 acetoxymethyl ester; ROS, reactive oxygen species; ΔΨm, mitochondrial membrane potential. MitoSOX and Mito Tracker Green; Invitrogen, Carlsbad, CA, USA.

Fig. 5 DEK increases nuclear translocation of Nrf2 and HO-1 expression. Effects of DEK on HO-1 expression (A) and nuclear Nrf2 translocation (B) were examined using Western blot analysis. HT22 neurons were exposed to different doses of DEK for 12 h. Total proteins were isolated for HO-1 and nuclear fractions were isolated at different time points for Nrf2. β-Actin and TBP were used as controls for equal protein loading in whole cells and nuclear fraction, respectively. Values were expressed as mean ± standard error of mean of four samples in one independent experiment. Statistical analyses were performed using one-way ANOVA followed by Bonferroni test. #p < 0.05, ##p < 0.01, and ###p < 0.001, as compared to untreated control group. DEK, dieckol; Nrf2, nuclear factor-like 2; HO-1, heme oxygenase-1; TBP, TATA binding protein.
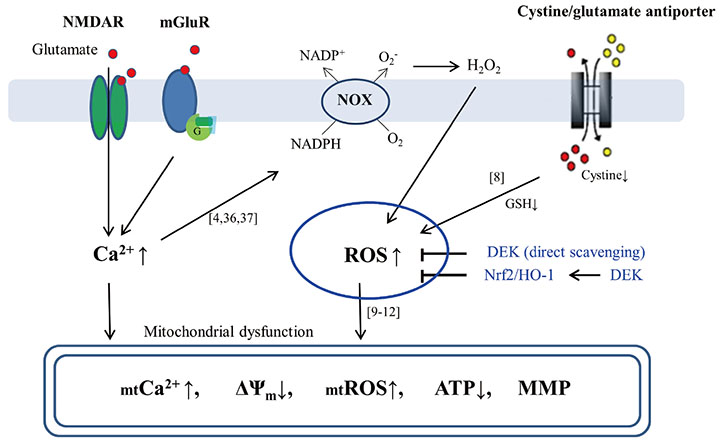
Fig. 6 Schematic diagram on neuroprotective effects of DEK against glutamate-induced neuronal cell death. There are two pathways underlying glutamate toxicity mechanism integrated in parallel in neurons; receptor-mediated excitotoxicity pathway and nonreceptor-mediated oxidative stress pathway. Increases of intracellular Ca2+ and ROS are triggering events in mitochondrial (mt) dysfunction and neuronal cell death against glutamate toxicity. Glutamate-induced intracellular Ca2+ increase is evoked mainly through NMDAR and G protein-coupled mGluR. Intracellular glutamate-induced ROS generation is induced through NOX and cystine/glutamate antiporters in the initial state. DEK exerted neuroprotective activities against glutamate toxicity through direct ROS scavenging and activation of the Nrf-2/HO-1 pathway as a cellular anti-oxidant defense system in this study. Reference numbers are shown in the known signaling pathways. NMDAR, N-methyl-D-aspartate receptors; mGluR, metabotropic glutamate receptors; NADPH, nicotinamide adenine dinuclelotide phosphate; NOX, NADPH oxidase; GSH, glutathione; ROS, reactive oxygen species; DEK, dieckol; Nrf2, nuclear factor-like 2; HO-1, heme oxygenase-1; ΔΨm, mitochondrial membrane potential; ATP, adenine triphosphate; MMP, mitochondrial membrane permeabilization.
Reference
-
1. Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988; 1:623–634.
Article2. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003; 4:552–565.
Article3. Duchen MR. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch. 2012; 464:111–121.
Article4. Ha JS, Lee JE, Lee JR, Lee CS, Maeng JS, Bae YS, Kwon KS, Park SS. Nox4-dependent H2O2 production contributes to chronic glutamate toxicity in primary cortical neurons. Exp Cell Res. 2010; 316:1651–1661.5. Peng TI, Jou MJ. Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci. 2010; 1201:183–188.
Article6. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989; 2:1547–1558.
Article7. Tobaben S, Grohm J, Seiler A, Conrad M, Plesnila N, Culmsee C. Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 2011; 18:282–292.
Article8. Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997; 19:453–463.
Article9. Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science. 2000; 290:1761–1765.
Article10. Pei Y, Xing D, Gao X, Liu L, Chen T. Real-time monitoring full length bid interacting with Bax during TNF-alpha-induced apoptosis. Apoptosis. 2007; 12:1681–1690.11. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007; 87:99–163.
Article12. Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999; 397:441–446.
Article13. Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prévost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000; 192:571–580.
Article14. Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002; 158:507–517.
Article15. Slemmer JE, Zhu C, Landshamer S, Trabold R, Grohm J, Ardeshiri A, Wagner E, Sweeney MI, Blomgren K, Culmsee C, Weber JT, Plesnila N. Causal role of apoptosis-inducing factor for neuronal cell death following traumatic brain injury. Am J Pathol. 2008; 173:1795–1805.
Article16. Valencia A, Sapp E, Kimm JS, McClory H, Reeves PB, Alexander J, Ansong KA, Masso N, Frosch MP, Kegel KB, Li X, DiFiglia M. Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington's disease. Hum Mol Genet. 2013; 22:1112–1131.
Article17. Cristóvão AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, Kim YS. NADPH oxidase 1 mediates α-synucleinopathy in Parkinson's disease. J Neurosci. 2012; 32:14465–14477.18. Choi BY, Jang BG, Kim JH, Lee BE, Sohn M, Song HK, Suh SW. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012; 1481:49–58.
Article19. Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999; 274:26071–26078.
Article20. Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003; 43:233–260.
Article21. Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem. 2005; 280:32485–32492.
Article22. Kim AR, Shin TS, Lee MS, Park JY, Park KE, Yoon NY, Kim JS, Choi JS, Jang BC, Byun DS, Park NK, Kim HR. Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and anti-inflammatory properties. J Agric Food Chem. 2009; 57:3483–3489.23. Koirala P, Jung HA, Choi JS. Recent advances in pharmacological research on Ecklonia species: a review. Arch Pharm Res. 2017; 40:981–1005.
Article24. Myung CS, Shin HC, Bao HY, Yeo SJ, Lee BH, Kang JS. Improvement of memory by dieckol and phlorofucofuroeckol in ethanol-treated mice: possible involvement of the inhibition of acetylcholinesterase. Arch Pharm Res. 2005; 28:691–698.
Article25. Cui Y, Park JY, Wu J, Lee JH, Yang YS, Kang MS, Jung SC, Park JM, Yoo ES, Kim SH, Ahn Jo S, Suk K, Eun SY. Dieckol attenuates microglia-mediated neuronal cell death via ERK, Akt and NADPH oxidase-mediated pathways. Korean J Physiol Pharmacol. 2015; 19:219–228.
Article26. Li Y, Qian ZJ, Ryu B, Lee SH, Kim MM, Kim SK. Chemical components and its antioxidant properties in vitro: an edible marine brown alga, Ecklonia cava. Bioorg Med Chem. 2009; 17:1963–1973.
Article27. Breyer A, Elstner M, Gillessen T, Weiser D, Elstner E. Glutamate-induced cell death in neuronal HT22 cells is attenuated by extracts from St. John's wort (Hypericum perforatum L.). Phytomedicine. 2007; 14:250–255.
Article28. Cui Y, Wu J, Jung SC, Park DB, Maeng YH, Hong JY, Kim SJ, Lee SR, Kim SJ, Kim SJ, Eun SY. Anti-neuroinflammatory activity of nobiletin on suppression of microglial activation. Biol Pharm Bull. 2010; 33:1814–1821.
Article29. Wu JJ, Cui Y, Yang YS, Jung SC, Hyun JW, Maeng YH, Park DB, Lee SR, Kim SJ, Eun SY. Mild mitochondrial depolarization is involved in a neuroprotective mechanism of Citrus sunki peel extract. Phytother Res. 2013; 27:564–571.30. Lee JH, Amarsanaa K, Wu J, Jeon SC, Cui Y, Jung SC, Park DB, Kim SJ, Han SH, Kim HW, Rhyu IJ, Eun SY. Nobiletin attenuates neurotoxic mitochondrial calcium overload through K+ influx and ΔΨm across mitochondrial inner membrane. Korean J Physiol Pharmacol. 2018; 22:311–319.31. Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006; 7:49.32. Noh HS, Hah YS, Nilufar R, Han J, Bong JH, Kang SS, Cho GJ, Choi WS. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J Neurosci Res. 2006; 83:702–709.
Article33. Kang TH, Bae KH, Yu MJ, Kim WK, Hwang HR, Jung H, Lee PY, Kang S, Yoon TS, Park SG, Ryu SE, Lee SC. Phosphoproteomic analysis of neuronal cell death by glutamate-induced oxidative stress. Proteomics. 2007; 7:2624–2635.
Article34. Azadmanesh J, Borgstahl GEO. A review of the catalytic mechanism of human manganese superoxide dismutase. Antioxidants (Basel). 2018; 7:pii: E25.
Article35. Chen C, Li L, Zhou HJ, Min W. The role of NOX4 and TRX2 in angiogenesis and their potential cross-talk. Antioxidants (Basel). 2017; 6:pii: E42.
Article36. Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009; 12:857–863.
Article37. Brennan-Minnella AM, Shen Y, El-Benna J, Swanson RA. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013; 4:e580.
Article38. Kwak JH, He Y, Yoon B, Koo S, Yang Z, Kang EJ, Lee BH, Han SY, Yoo YC, Lee KB, Kim JS. Synthesis of rhodamine-labelled dieckol: its unique intracellular localization and potent anti-inflammatory activity. Chem Commun (Camb). 2014; 50:13045–13048.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Fraxetin Induces Heme Oxygenase-1 Expression by Activation of Akt/Nrf2 or AMP-activated Protein Kinase α/Nrf2 Pathway in HaCaT Cells
- Cytoprotective Effects of Serum Hormone Deprivation against Glutamate Toxicity in HT22 Mouse Hippocampal Cells
- Downregulation of Reactive Oxygen Species in Apoptosis
- SiO2 Nanoparticles Induced Cytotoxicity by Oxidative Stress in Human Bronchial Epithelial Cell, Beas-2B
- Neuroprotective Effect of β-Lapachone against Glutamate-Induced Injury in HT22 Cells
