Anti-SEMA3A Antibody: A Novel Therapeutic Agent to Suppress Glioblastoma Tumor Growth
- Affiliations
-
- 1Institute for Refractory Cancer Research, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. nsnam@skku.edu
- 2Department of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology (SAIHST), Sungkyunkwan University, Seoul, Korea.
- 3Department of Neurosurgery, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- 4Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- 5Department of Stem Cell Biology and Regenerative Medicine, Lerner Research Institute, Cleveland Clinic, Cleveland, OH, USA.
- KMID: 2417889
- DOI: http://doi.org/10.4143/crt.2017.315
Abstract
- PURPOSE
Glioblastoma (GBM) is classified as one of the most aggressive and lethal brain tumor. Great strides have been made in understanding the genomic and molecular underpinnings of GBM, which translated into development of new therapeutic approaches to combat such deadly disease. However, there are only few therapeutic agents that can effectively inhibit GBM invasion in a clinical framework. In an effort to address such challenges, we have generated anti-SEMA3A monoclonal antibody as a potential therapeutic antibody against GBM progression.
MATERIALS AND METHODS
We employed public glioma datasets, Repository of Molecular Brain Neoplasia Data and The Cancer Genome Atlas, to analyze SEMA3A mRNA expression in human GBM specimens. We also evaluated for protein expression level of SEMA3A via tissue microarray (TMA) analysis. Cell migration and proliferation kinetics were assessed in various GBM patient-derived cells (PDCs) and U87-MG cell-line for SEMA3A antibody efficacy. GBM patient-derived xenograft (PDX) models were generated to evaluate tumor inhibitory effect of anti-SEMA3A antibody in vivo.
RESULTS
By combining bioinformatics and TMA analysis, we discovered that SEMA3A is highly expressed in human GBM specimens compared to non-neoplastic tissues. We developed three different anti-SEMA3A antibodies, in fully human IgG form, through screening phage-displayed synthetic antibody library using a classical panning method. Neutralization of SEMA3A significantly reduced migration and proliferation capabilities of PDCs and U87-MG cell line in vitro. In PDX models, treatment with anti-SEMA3A antibody exhibited notable tumor inhibitory effect through down-regulation of cellular proliferative kinetics and tumor-associated macrophages recruitment.
CONCLUSION
In present study, we demonstrated tumor inhibitory effect of SEMA3A antibody in GBM progression and present its potential relevance as a therapeutic agent in a clinical framework.
MeSH Terms
Figure
-

Fig. 1. SEMA3A is highly expressed in glioblastoma (GBM) specimens compared to the normal brains. (A) REMBRANDT microarray data analysis for SEMA3A mRNA expression levels corresponding to glioma grades II-IV. WHO, World Health Organization. ***p < 0.001. (B) Kaplan-Meier survival curves of GBM patient corresponding to SEMA3A expression levels in The Cancer Genome Atlas datasets. (C) Immunohistochemistry of SEMA3A on tissue microarray. (D) Representative analysis of SEMA3A expressions in 27 GBM paired tumor and normal specimens.

Fig. 2. Identification of SEMA3A-specific single chain fragment variant (scFv) using synthetic phage library. (A) The efficacy of classical panning was evaluated by the ratio of output phage titer to input phage titer. (B) Based on enzyme-linked immunosorbent assay (ELISA) results, 52 positive clones specific to human SEMA3A were selected from among 86 clones which were selected randomly in the fourth round of the output mixture. (C) The three scFvs purified using Ni-NTA resin and poly-column were eluted by phosphate buffered saline with 200 mM imidazole (pH7.4). Molecular sizes of the three anti-SEMA3A clones (A08, C10, and F11) were verified by Coomassie blue staining. (D) hSEMA3A binding activities of three anti-SEMA antibodies and 12B (a negative control) were confirmed by indirect ELISA. 96-well plates coated with hSEMA3A (1 μg/mL) or bovine serum albumin (BSA; 1 μg/mL) were bound for three anti-SEMA3A antibodies and 12B. Then, antihemaglutinin antibody conjugated to horseradish peroxidase (HRP) was used for bound scFvs detection. The absorbance of each well for HRP reactions was measured at 450 nm. OD, optical density.
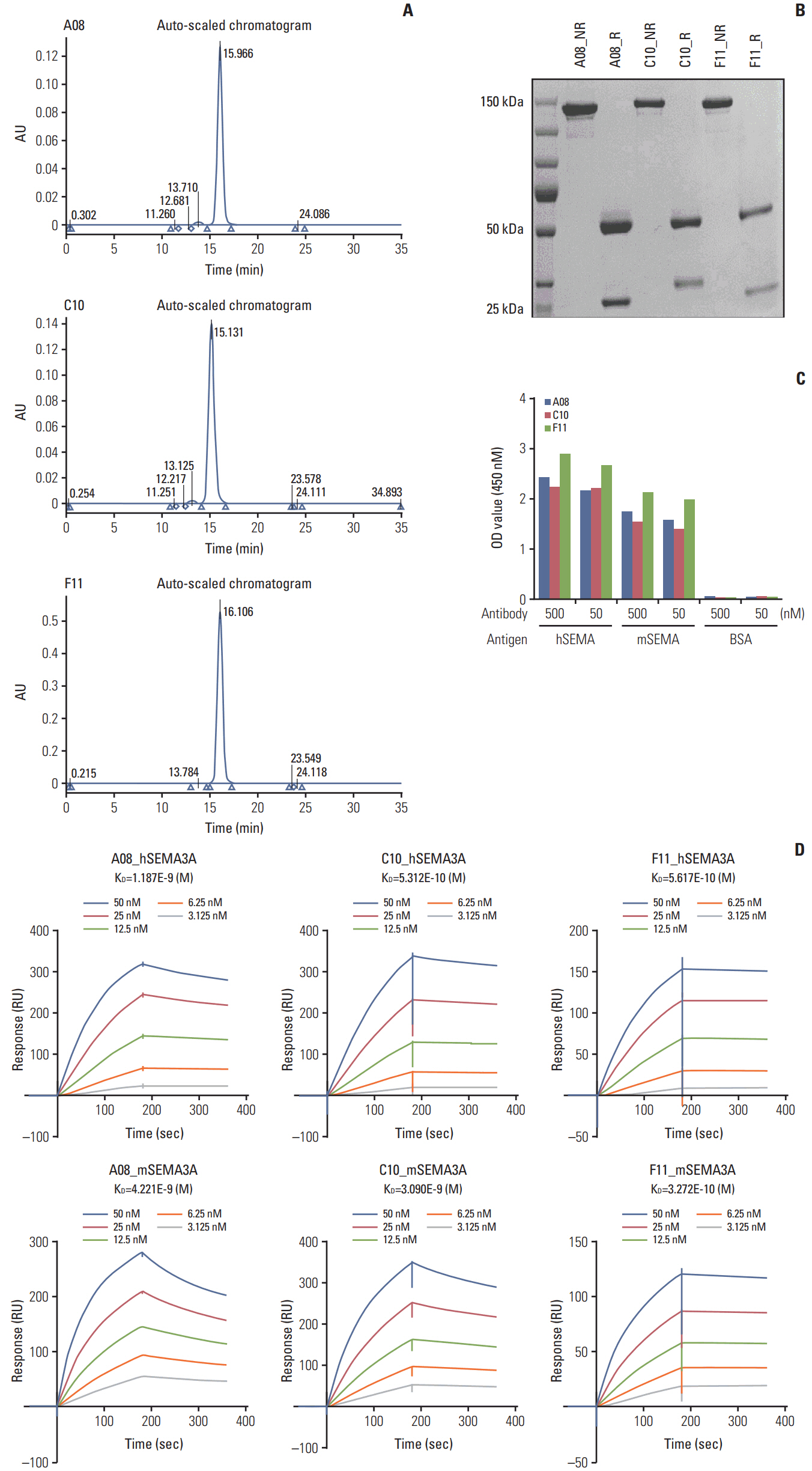
Fig. 3. Production of anti-SEMA3A IgG antibodies. (A) Purity of three anti-SEMA3A IgGs determined by high performance liquid chromatography. The peak at 15-16 minutes indicates γ-globulin (~150 kDa). The purity of all three anti-SEMA3A IgGs was over 98%. (B) Sizes of the anti-SEMA3A IgGs were verified in non-reducing (NR) and reducing (R) conditions by sodium dodecyl sulfate polyacrylamide gel electrophoresis. In NR condition, fully IgG was detected whereas in R condition where cleavages disulfide bond heavy chain and light chain were observed. (C) To determinate binding activity of human and mouse SEMA3A, three anti-SEMA3A IgGs were analyzed on enzyme-linked immunosorbent assay (ELISA) plates when the presence of hSEMA3A, mSEMA3A, and bovine serum albumin (BSA) as negative control. Optical density (OD) values derived from indirect ELISA in which SEMA3A IgG was captured via anti-human Fab antibody conjugated to horseradish peroxidase. (D) Use of the BIAcore to determine the calculated KD value of three anti-SEMA3A IgGs to human and mouse SEMA3A. The assay method was Fab-based capture format via human Fab capture kit (GE Healthcare).
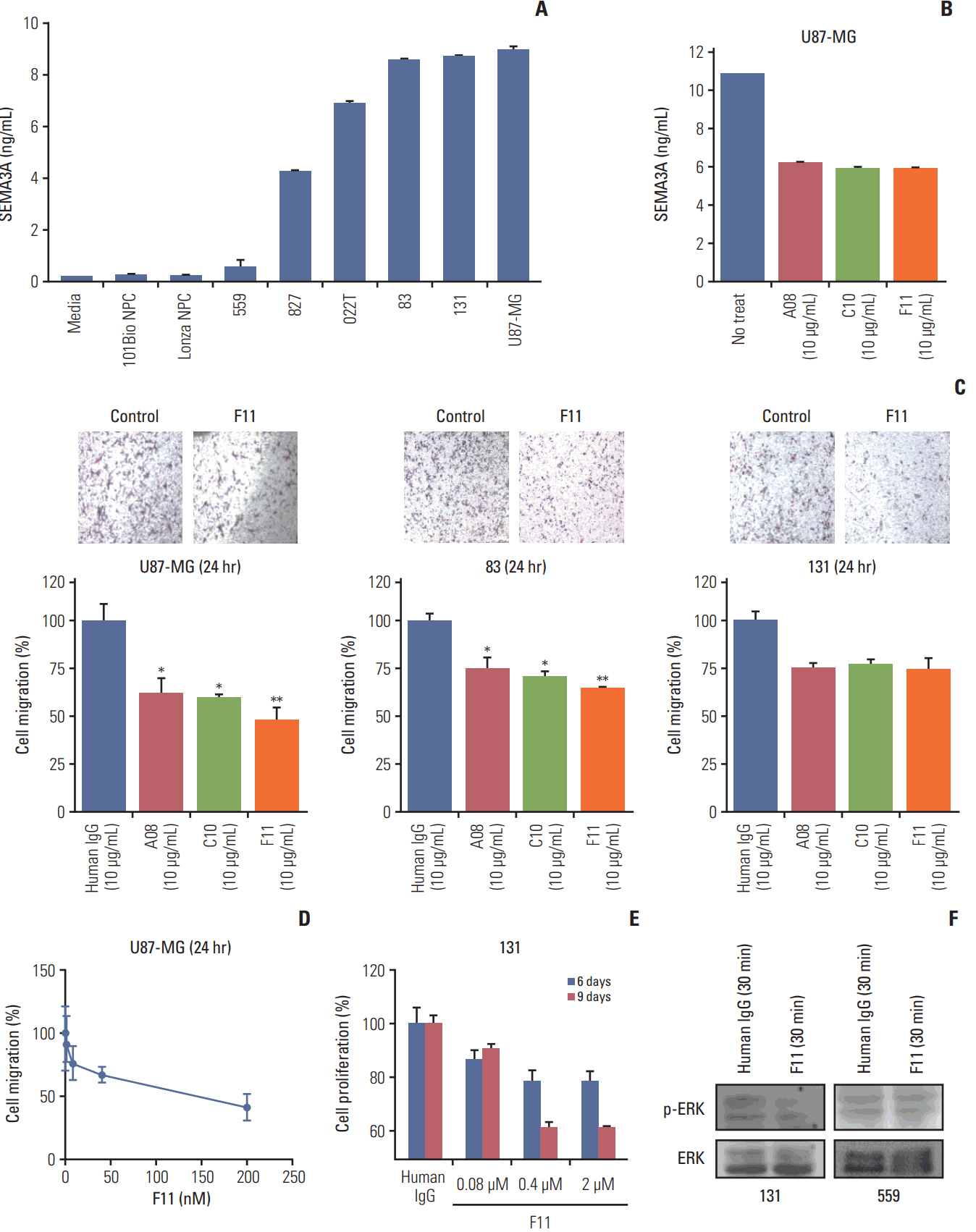
Fig. 4. Anti-SEMA3A IgG antibody impedes migration and proliferation of patient-derived glioblastoma (GBM) cells. (A) The concentration of SEMA3A secreted for a day was measured by sandwich enzyme-linked immunosorbent assay (ELISA) in various cell types. GBM patient cells and U87-MG cells as a positive control and neural progenitor cell and medium as negative controls. (B) Neutralization assay of three anti-SEMA3A IgGs in U87-MG cells. The evaluate method is same method for U87-MG cells were incubated with anti-SEMA3A IgG and the amount of SEMA3A in medium was measured by sandwich ELISA. (C) While the migration rate of cells (U87-MG, 131, and 83) treated with control human IgG remains unchanged, migration rates of cells treated with anti-SEMA3A IgGs for 1 day were impeded significantly. Each antibody treatment concentration was 10 μg/mL and macroscopic observation of the transwell chambers. *p < 0.05, **p < 0.01. (D) Inhibition of migration activity by anti-SEMA3A F11 for one day was evaluated using Oris cell migration assay. The level of migration activity inhibition was proportional to the concentration gradient of anti-SEMA3A F11. Migration activity was reduced by 52% compared to the control IgG-treated cells in the max concentration of F11 (200 nM) treated cells. (E) Comparison of the effect of anti-SEMA3A F11 and control human IgG (2 μM) on in-vitro proliferation of 131 cells. On the 9 days after F11 treatment, 131 proliferation was inhibited by 60% of control human IgG treated cells. (F) Immunoblots of phospho-ERK and ERK in control human IgG or F11 IgG treated 131 and 559 cells. The SEMA3A hypersecreting 131 cells decreased ERK phosphorylation during F11 treatment and remained unchanged for ERK phosphorylation of 559 cells that secretes less SEMA3A.
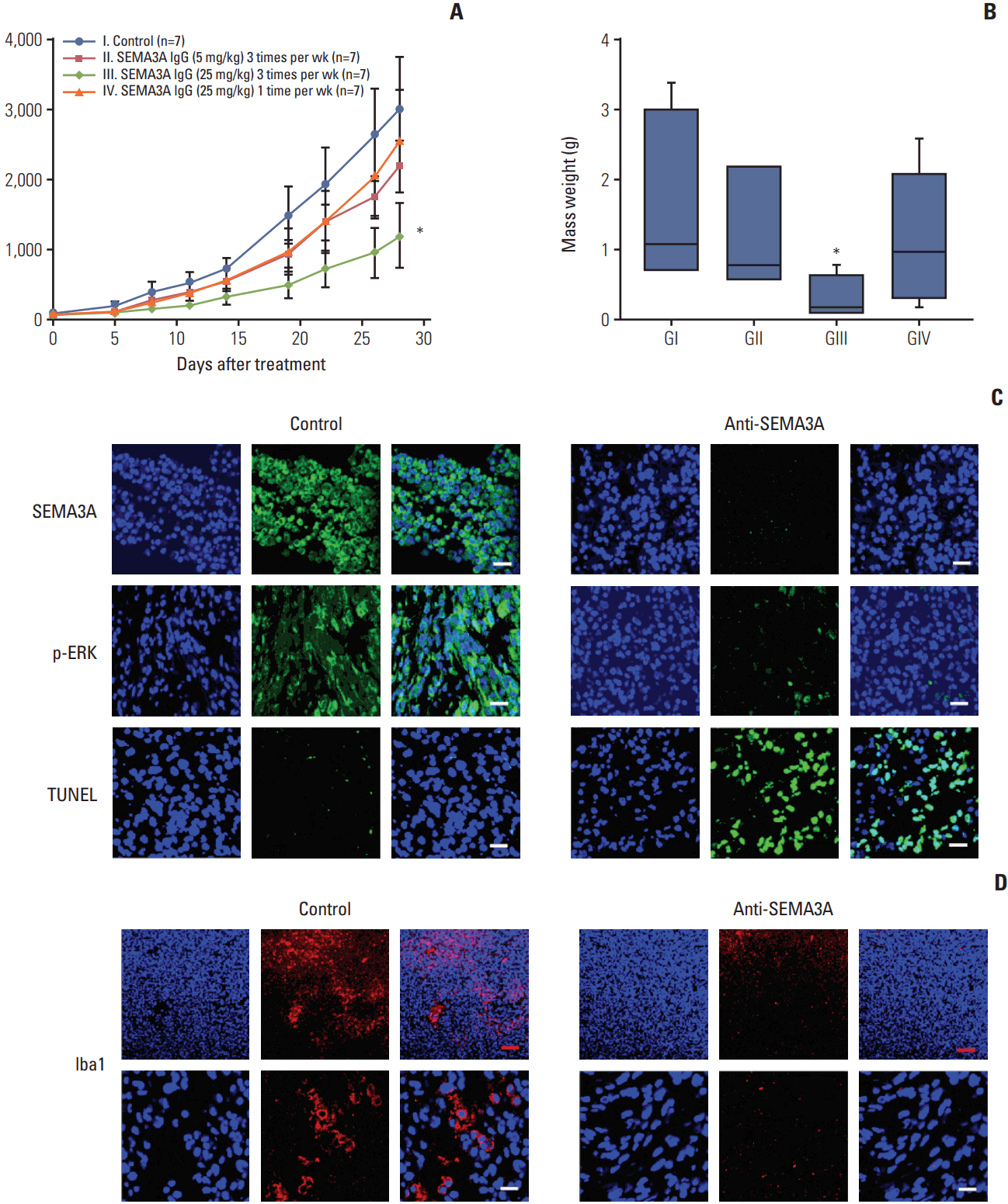
Fig. 5. In vivo effects of anti-SEMA3A antibody on the glioblastoma (GBM) tumor growth. (A) Antitumor activity of antiSEMA3A F11 in GBM patient cell 131 xenograft model. When tumors were approximately 80 mm3 , the patient-derived xenograft tumors were randomly assigned into study groups (n=7 mice/group) and were participated in the dosing phase of the study. Treatment of F11 was administered intravenously at 5 mg/kg and 25 mg/kg with all treatments well tolerated and anti-SEMA3A F11 injections were stopped at day 28. *p < 0.05. (B) Mass weight variation of mice was similar to mass size affected by anti-SEMA3A F11 within groups. *p < 0.05. (C, D) Representative immunofluorescence images of SEMA3A, p-ERK, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and Iba1 staining using paraffin sections of tumors. Scale bars=20 μm (white), 100 μm (red).
Reference
-
References
1. Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, et al. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs. 2009; 18:1061–83.
Article2. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
Article3. Reardon DA, Wen PY. Therapeutic advances in the treatment of glioblastoma: rationale and potential role of targeted agents. Oncologist. 2006; 11:152–64.
Article4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–96.
Article5. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008; 100:35–83.
Article6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74.
Article7. Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012; 26:756–84.
Article8. Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer. 2001; 8:161–73.
Article9. Herbst RS, Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002; 94:1593–611.10. Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013; 341:1192–8.
Article11. Tamagnone L, Comoglio PM. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol. 2000; 10:377–83.
Article12. Kruger RP, Aurandt J, Guan KL. Semaphorins command cells to move. Nat Rev Mol Cell Biol. 2005; 6:789–800.
Article13. Neufeld G, Sabag AD, Rabinovicz N, Kessler O. Semaphorins in angiogenesis and tumor progression. Cold Spring Harb Perspect Med. 2012; 2:a006718.
Article14. Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993; 75:217–27.
Article15. Kigel B, Varshavsky A, Kessler O, Neufeld G. Successful inhibition of tumor development by specific class-3 semaphorins is associated with expression of appropriate semaphorin receptors by tumor cells. PLoS One. 2008; 3:e3287.
Article16. Acevedo LM, Barillas S, Weis SM, Gothert JR, Cheresh DA. Semaphorin 3A suppresses VEGF-mediated angiogenesis yet acts as a vascular permeability factor. Blood. 2008; 111:2674–80.
Article17. Muller MW, Giese NA, Swiercz JM, Ceyhan GO, Esposito I, Hinz U, et al. Association of axon guidance factor semaphorin 3A with poor outcome in pancreatic cancer. Int J Cancer. 2007; 121:2421–33.18. Bagci T, Wu JK, Pfannl R, Ilag LL, Jay DG. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene. 2009; 28:3537–50.
Article19. Treps L, Edmond S, Harford-Wright E, Galan-Moya EM, Schmitt A, Azzi S, et al. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene. 2016; 35:2615–23.
Article20. Hu ZQ, Zhou SL, Zhou ZJ, Luo CB, Chen EB, Zhan H, et al. Overexpression of semaphorin 3A promotes tumor progression and predicts poor prognosis in hepatocellular carcinoma after curative resection. Oncotarget. 2016; 7:51733–46.
Article21. Kretzschmar T, von Ruden T. Antibody discovery: phage display. Curr Opin Biotechnol. 2002; 13:598–602.
Article22. Hoogenboom HR, de Bruine AP, Hufton SE, Hoet RM, Arends JW, Roovers RC. Antibody phage display technology and its applications. Immunotechnology. 1998; 4:1–20.
Article23. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006; 9:391–403.
Article24. Yang HY, Kang KJ, Chung JE, Shim H. Construction of a large synthetic human scFv library with six diversified CDRs and high functional diversity. Mol Cells. 2009; 27:225–35.
Article25. Shinohara H, Yagita H, Ikawa Y, Oyaizu N. Fas drives cell cycle progression in glioma cells via extracellular signal-regulated kinase activation. Cancer Res. 2000; 60:1766–72.26. Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013; 24:695–709.
Article27. Rivera LB, Bergers G. Location, location, location: macrophage positioning within tumors determines pro- or antitumor activity. Cancer Cell. 2013; 24:687–9.
Article28. Solinas G, Germano G, Mantovani A, Allavena P. Tumorassociated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009; 86:1065–73.
Article29. Cerani A, Tetreault N, Menard C, Lapalme E, Patel C, Sitaras N, et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab. 2013; 18:505–18.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Expression of Semaphorin 3A and Neuropilin 1 in Asthma
- Expression of Epidermal Growth Factor Receptor in Astrocytoma
- Netrin-1 Specifically Enhances Cell Spreading on Fibronectin in Human Glioblastoma Cells
- Anti-Migration and Anti-Invasion Effects of Curcumin via Suppression of Fascin Expression in Glioblastoma Cells
- Visualization of Tumor Angiogenesis Using MR Imaging Contrast Agent Gd-DTPA-anti-VEGF Receptor 2 Antibody Conjugate in a Mouse Tumor Model
