Primary Carnitine Deficiency and Cardiomyopathy
- Affiliations
-
- 1Department of Cardiology, Shanghai Children's Medical Center, Shanghai Jiaotong University School of Medicine, Shanghai, China. chensb@sh163.net
- KMID: 2224771
- DOI: http://doi.org/10.4070/kcj.2013.43.12.785
Abstract
- Carnitine is essential for the transfer of long-chain fatty acids from the cytosol into mitochondria for subsequent beta-oxidation. A lack of carnitine results in impaired energy production from long-chain fatty acids, especially during periods of fasting or stress. Primary carnitine deficiency (PCD) is an autosomal recessive disorder of mitochondrial beta-oxidation resulting from defective carnitine transport and is one of the rare treatable etiologies of metabolic cardiomyopathies. Patients affected with the disease may present with acute metabolic decompensation during infancy or with severe cardiomyopathy in childhood. Early recognition of the disease and treatment with L-carnitine may be life-saving. In this review article, the pathophysiology, clinical presentation, diagnosis, treatment and prognosis of PCD are discussed, with a focus on cardiac involvements.
MeSH Terms
Figure
-
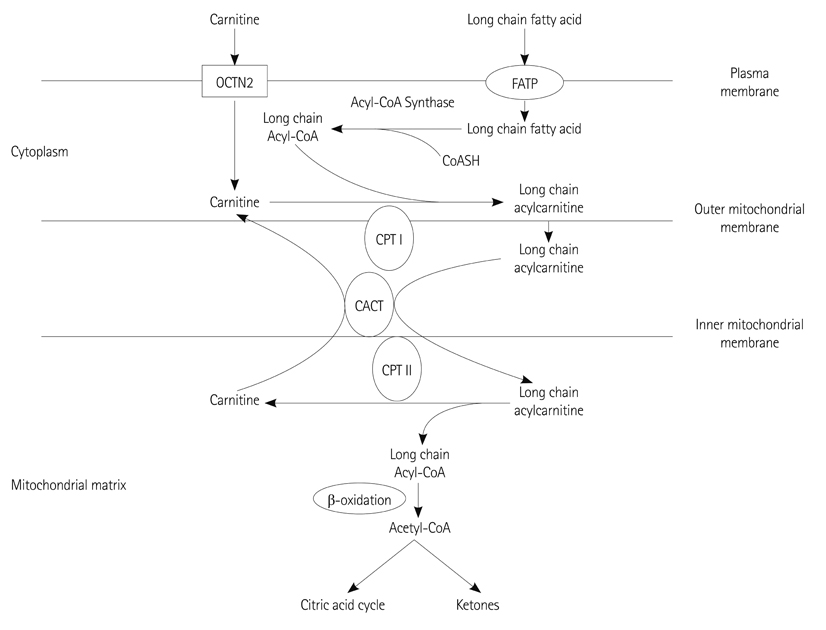
Fig. 1 The role of carnitine in the transport of mitochondrial long-chain fatty acid oxidation. Carnitine is actively transported via OCTN2 into the cytoplasm and forms ester bonds with long chain carboxylic acids by the action of carnitine palmitoyl transferase I (CPT I), located in the inner aspect of the outer mitochondrial membrane. Acylcarnitine is then translocated across the inner mitochondrial membrane by the carnitine acylcarnitine translocase (CACT) and cleaved by CPT II in the inner aspect of the inner mitochondrial membrane. Carnitine is released in the mitochondrial matrix and then return to the cytoplasm for another cycle, while the fatty acids are conjugated back to coenzyme A in the mitochondrial matrix and then enter the pathway of β-oxidation and ketone synthesis. FATP: fatty acid transport protein.
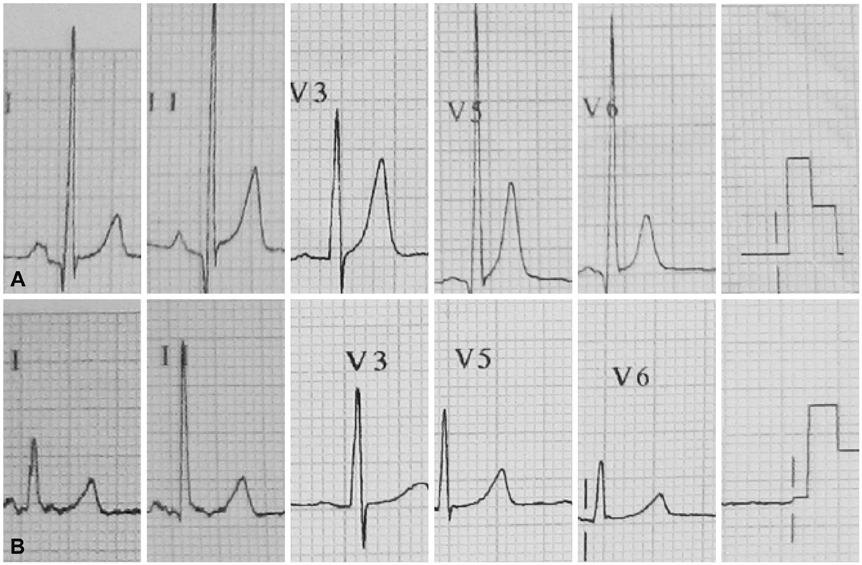
Fig. 2 Electrocardiographic tracing before and after treatment with L-carnitine. A: before treatment, showing a shortened QT interval (QTc 330 msec) associated with an abnormally tall and symmetrical peaking T wave. B: after one-month L-carnitine therapy, T-wave amplitude and QT interval (QTc 401 msec) return to normal.
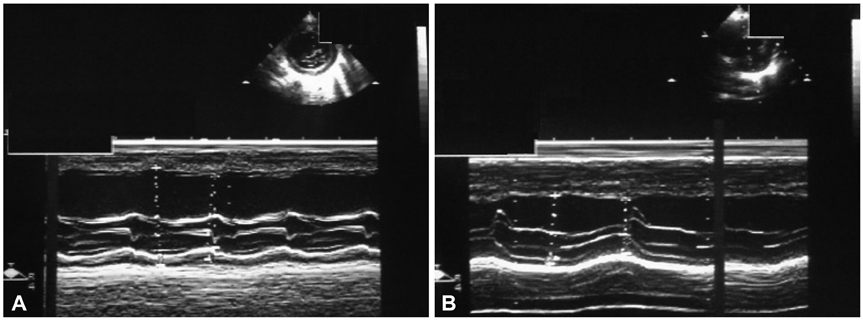
Fig. 3 Transthoracic echocardiography before and after treatment with L-carnitine. A: M-mode echocardiogram demonstrating a severely enlarged left ventricle (LVEDD 5.40 cm) with poor systolic function (LVEF 40.6%) on initial examination (study). B: after five-month L-carnitine therapy, the left ventricle return to normal size (LVEDD 3.09 cm) and systolic function (LVEF 65.9%). LVEF: left ventricular ejection fraction, LVEDD: left ventricular end-diastolic diameter.
Reference
-
1. Cox GF, Sleeper LA, Lowe AM, et al. Factors associated with establishing a causal diagnosis for children with cardiomyopathy. Pediatrics. 2006; 118:1519–1531.2. Cox GF. Diagnostic Approaches to Pediatric Cardiomyopathy of Metabolic Genetic Etiologies and Their Relation to Therapy. Prog Pediatr Cardiol. 2007; 24:15–25.3. Tripp ME, Katcher ML, Peters HA, et al. Systemic carnitine deficiency presenting as familial endocardial fibroelastosis: a treatable cardiomyopathy. N Engl J Med. 1981; 305:385–390.4. Waber LJ, Valle D, Neill C, DiMauro S, Shug A. Carnitine deficiency presenting as familial cardiomyopathy: a treatable defect in carnitine transport. J Pediatr. 1982; 101:700–705.5. Pierpont ME, Breningstall GN, Stanley CA, Singh A. Familial carnitine transporter defect: a treatable cause of cardiomyopathy in children. Am Heart J. 2000; 139(2 Pt 3):S96–S106.6. Rebouche CJ. Carnitine function and requirements during the life cycle. FASEB J. 1992; 6:3379–3386.7. Glube N, Closs E, Langguth P. OCTN2-mediated carnitine uptake in a newly discovered human proximal tubule cell line (Caki-1). Mol Pharm. 2007; 4:160–168.8. Flanagan JL, Simmons PA, Vehige J, Willcox MD, Garrett Q. Role of carnitine in disease. Nutr Metab (Lond). 2010; 7:30.9. Koizumi A, Nozaki J, Ohura T, et al. Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet. 1999; 8:2247–2254.10. Lund AM, Joensen F, Hougaard DM, et al. Carnitine transporter and holocarboxylase synthetase deficiencies in The Faroe Islands. J Inherit Metab Dis. 2007; 30:341–349.11. Rasmussen J, Nielsen OW, Janzen N, et al. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the Faroe Islands. J Inherit Metab Dis. 2013; [Epub ahead of print].12. Nezu J, Tamai I, Oku A, et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet. 1999; 21:91–94.13. Wang Y, Ye J, Ganapathy V, Longo N. Mutations in the organic cation/carnitine transporter OCTN2 in primary carnitine deficiency. Proc Natl Acad Sci U S A. 1999; 96:2356–2360.14. Tamai I, Ohashi R, Nezu J, et al. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998; 273:20378–20382.15. Wu X, Prasad PD, Leibach FH, Ganapathy V. cDNA sequence, transport function, and genomic organization of human OCTN2, a new member of the organic cation transporter family. Biochem Biophys Res Commun. 1998; 246:589–595.16. Scaglia F, Wang Y, Singh RH, et al. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet Med. 1998; 1:34–39.17. Stanley CA, Bennett MJ. Defects in Metabolism of Lipid. In : Kliegman RM, Behrman RE, Stanton BF, St. Geme J, Schor N, editors. Nelson textbook of pediatrics. 19th ed. Philadelphia: W.B. Saunders Company;2011. p. 460–461.18. Paulson DJ. Carnitine deficiency-induced cardiomyopathy. Mol Cell Biochem. 1998; 180:33–41.19. Chapoy PR, Angelini C, Brown WJ, Stiff JE, Shug AL, Cederbaum SD. Systemic carnitine deficiency--a treatable inherited lipid-storage disease presenting as Reye's syndrome. N Engl J Med. 1980; 303:1389–1394.20. Shibbani K, Fahed A, Al-Shaar L, et al. Primary carnitine deficiency: novel mutations and insights into the cardiac phenotype. Clin Genet. 2013; [Epub ahead of print].21. Kilic M, Ozgül RK, Coşkun T, et al. Identification of mutations and evaluation of cardiomyopathy in Turkish patients with primary carnitine deficiency. JIMD Rep. 2012; 3:17–23.22. Fu LJ, Chen SB, Han LS, et al. [Clinical presentation and therapeutic outcomes of carnitine deficiency-induced cardiomyopathy]. Zhonghua Er Ke Za Zhi. 2012; 50:929–934.23. Stanley CA, DeLeeuw S, Coates PM, et al. Chronic cardiomyopathy and weakness or acute coma in children with a defect in carnitine uptake. Ann Neurol. 1991; 30:709–716.24. Rasmussen J, Køber L, Lund AM, Nielsen OW. Primary Carnitine deficiency in the Faroe Islands: health and cardiac status in 76 adult patients diagnosed by screening. J Inherit Metab Dis. 2013; [Epub ahead of print].25. Lee NC, Tang NL, Chien YH, et al. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol Genet Metab. 2010; 100:46–50.26. De Biase I, Champaigne NL, Schroer R, Pollard LM, Longo N, Wood T. Primary Carnitine Deficiency Presents Atypically with Long QT Syndrome: A Case Report. JIMD Rep. 2012; 2:87–90.27. Amat di San Filippo C, Taylor MR, Mestroni L, Botto LD, Longo N. Cardiomyopathy and carnitine deficiency. Mol Genet Metab. 2008; 94:162–166.28. Labarthe F, Benoist JF, Peralta M, et al. Primary carnitine uptake deficiency is associated with short QT syndrome and ventricular fibrillation. 11th International Congress of Inborn errors of Metabolism, San Diego CA. Mol Genet Metab. 2009; 98:55–56. (Poster 327).29. Rijlaarsdam RS, van Spronsen FJ, Bink-Boelkens MT, et al. Ventricular fibrillation without overt cardiomyopathy as first presentation of organic cation transporter 2-deficiency in adolescence. Pacing Clin Electrophysiol. 2004; 27:675–676.30. El-Hattab AW, Li FY, Shen J, et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: clinical, biochemical, and molecular aspects. Genet Med. 2010; 12:19–24.31. Rinaldo P, Stanley CA, Hsu BY, Sanchez LA, Stern HJ. Sudden neonatal death in carnitine transporter deficiency. J Pediatr. 1997; 131:304–305.32. Rasmussen J, Nielsen OW, Lund AM, Køber L, Djurhuus H. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J Inherit Metab Dis. 2013; 36:35–41.33. Matsuishi T, Hirata K, Terasawa K, et al. Successful carnitine treatment in two siblings having lipid storage myopathy with hypertrophic cardiomyopathy. Neuropediatrics. 1985; 16:6–12.34. Tein I, De Vivo DC, Bierman F, et al. Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy. Pediatr Res. 1990; 28:247–255.35. Ferro F, Ouillé A, Tran TA, et al. Long-chain acylcarnitines regulate the hERG channel. PLoS One. 2012; 7:e41686.36. Garavaglia B, Uziel G, Dworzak F, Carrara F, DiDonato S. Primary carnitine deficiency: heterozygote and intrafamilial phenotypic variation. Neurology. 1991; 41:1691–1693.37. Xiaofei E, Wada Y, Dakeishi M, et al. Age-associated cardiomyopathy in heterozygous carrier mice of a pathological mutation of carnitine transporter gene, OCTN2. J Gerontol A Biol Sci Med Sci. 2002; 57:B270–B278.38. Takahashi R, Asai T, Murakami H, et al. Pressure overload-induced cardiomyopathy in heterozygous carrier mice of carnitine transporter gene mutation. Hypertension. 2007; 50:497–502.39. Sarafoglou K, Tridgell AH, Bentler K, Redlinger-Grosse K, Berry SA, Schimmenti LA. Cardiac conduction improvement in two heterozygotes for primary carnitine deficiency on L-carnitine supplementation. Clin Genet. 2010; 78:191–194.40. Wang Y, Taroni F, Garavaglia B, Longo N. Functional analysis of mutations in the OCTN2 transporter causing primary carnitine deficiency: lack of genotype-phenotype correlation. Hum Mutat. 2000; 16:401–407.41. Spiekerkoetter U, Huener G, Baykal T, et al. Silent and symptomatic primary carnitine deficiency within the same family due to identical mutations in the organic cation/carnitine transporter OCTN2. J Inherit Metab Dis. 2003; 26:613–615.42. Rose EC, di San Filippo CA, Ndukwe Erlingsson UC, Ardon O, Pasquali M, Longo N. Genotype-phenotype correlation in primary carnitine deficiency. Hum Mutat. 2012; 33:118–123.43. Yamak A, Bitar F, Karam P, Nemer G. Exclusive cardiac dysfunction in familial primary carnitine deficiency cases: a genotype-phenotype correlation. Clin Genet. 2007; 72:59–62.44. Magoulas PL, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2012; 7:68.45. Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006; 142C:77–85.46. Helton E, Darragh R, Francis P, et al. Metabolic aspects of myocardial disease and a role for L-carnitine in the treatment of childhood cardiomyopathy. Pediatrics. 2000; 105:1260–1270.47. Kothari SS, Sharma M. L-carnitine in children with idiopathic dilated cardiomyopathy. Indian Heart J. 1998; 50:59–61.48. Rizos I. Three-year survival of patients with heart failure caused by dilated cardiomyopathy and L-carnitine administration. Am Heart J. 2000; 139(2 Pt 3):S120–S123.49. Agnetti A, Bitton L, Tchana B, Raymond A, Carano N. Primary carnitine deficiency dilated cardiomyopathy: 28 years follow-up. Int J Cardiol. 2013; 162:e34–e35.50. Cederbaum SD, Koo-McCoy S, Tein I, et al. Carnitine membrane transporter deficiency: a long-term follow up and OCTN2 mutation in the first documented case of primary carnitine deficiency. Mol Genet Metab. 2002; 77:195–201.51. Stanley CA. Carnitine deficiency disorders in children. Ann N Y Acad Sci. 2004; 1033:42–51.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- The effects of L-carnitine in congestive cardiomyopathy
- Systemic primary carnitine deficiency with hypoglycemic encephalopathy
- A Study for the Normal Serum Carnitine Levels and the Effect of Anticonvulsants on Serum Carnitine Levels in Pediatric Age
- L-carnitine in maintenance hemodialysis clinical, lipid and biochemical effects
- A Case of Pulmonary Embolism with Protein C, S Deficiency in Apical Hypertrophic Cardiomyopathy
