Integrin αvβ3 Induces HSP90 Inhibitor Resistance via FAK Activation in KRAS-Mutant Non-Small Cell Lung Cancer
- Yoon S
 1
1 - Yang H2
- Ryu HM1,3
- Lee E1,3
- Jo Y1
- Seo S1
- Kim D4
- Lee CH5
- Kim W6
- Jung KH1
- Park SR1
- Choi EK7,8
- Kim SW1
- Park KS1,3
- Lee DH1
- Affiliations
-
- 1Department of Oncology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- 2Division of Medical Oncology, Department of Internal Medicine, CHA Bundang Medical Center, CHA University, Seongnam, Korea
- 3Department of Biomedical Sciences, University of Ulsan College of Medicine, Asan Medical Center, Seoul, Korea
- 4Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- 5Bio & Drug Discovery Division, Center for Drug Discovery Technology, Korea Research Institute of Chemical Technology, Daejeon, Korea
- 6Department of Orthopaedic Surgery, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- 7Center for Advancing Cancer Therapeutics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- 8Department of Radiation Oncology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- KMID: 2531323
- DOI: http://doi.org/10.4143/crt.2021.651
Abstract
- Purpose
Heat shock protein-90 (HSP90) remains an important cancer target because of its involvement in multiple oncogenic protein pathways and biologic processes. Although many HSP90 inhibitors have been tested in the treatment of KRAS-mutant non–small cell lung cancer (NSCLC), most, including AUY922, have failed due to toxic effects and resistance generation, even though a modest efficacy has been observed for these drugs in clinical trials. In our present study, we investigated the novel mechanism of resistance to AUY922 to explore possible avenues of overcoming and want to provide some insights that may assist with the future development of successful next-generation HSP90 inhibitors.
Materials and Methods
We established two AUY922-resistant KRAS-mutated NSCLC cells and conducted RNA sequencing to identify novel resistance biomarker.
Results
We identified novel two resistance biomarkers. We observed that both integrin Av (ITGAv) and β3 (ITGB3) induce AUY922-resistance via focal adhesion kinase (FAK) activation, as well as an epithelial-mesenchymal transition, in both in vitro and in vivo xenograft model. mRNAs of both ITGAv and ITGB3 were also found to be elevated in a patient who had shown acquired resistance in a clinical trial of AUY922. ITGAv was induced by miR-142 downregulation, and ITGB3 was increased by miR-150 downregulation during the development of AUY922-resistance. Therefore, miR-150 and miR-142 overexpression effectively inhibited ITGAvB3-dependent FAK activation, restoring sensitivity to AUY922.
Conclusion
The synergistic co-targeting of FAK and HSP90 attenuated the growth of ITGAvB3-induced AUY922-resistant KRAS-mutated NSCLC cells in vitro and in vivo, suggesting that this combination may overcome acquired AUY922-resistance in KRAS-mutant NSCLC.
Keyword
Figure
-
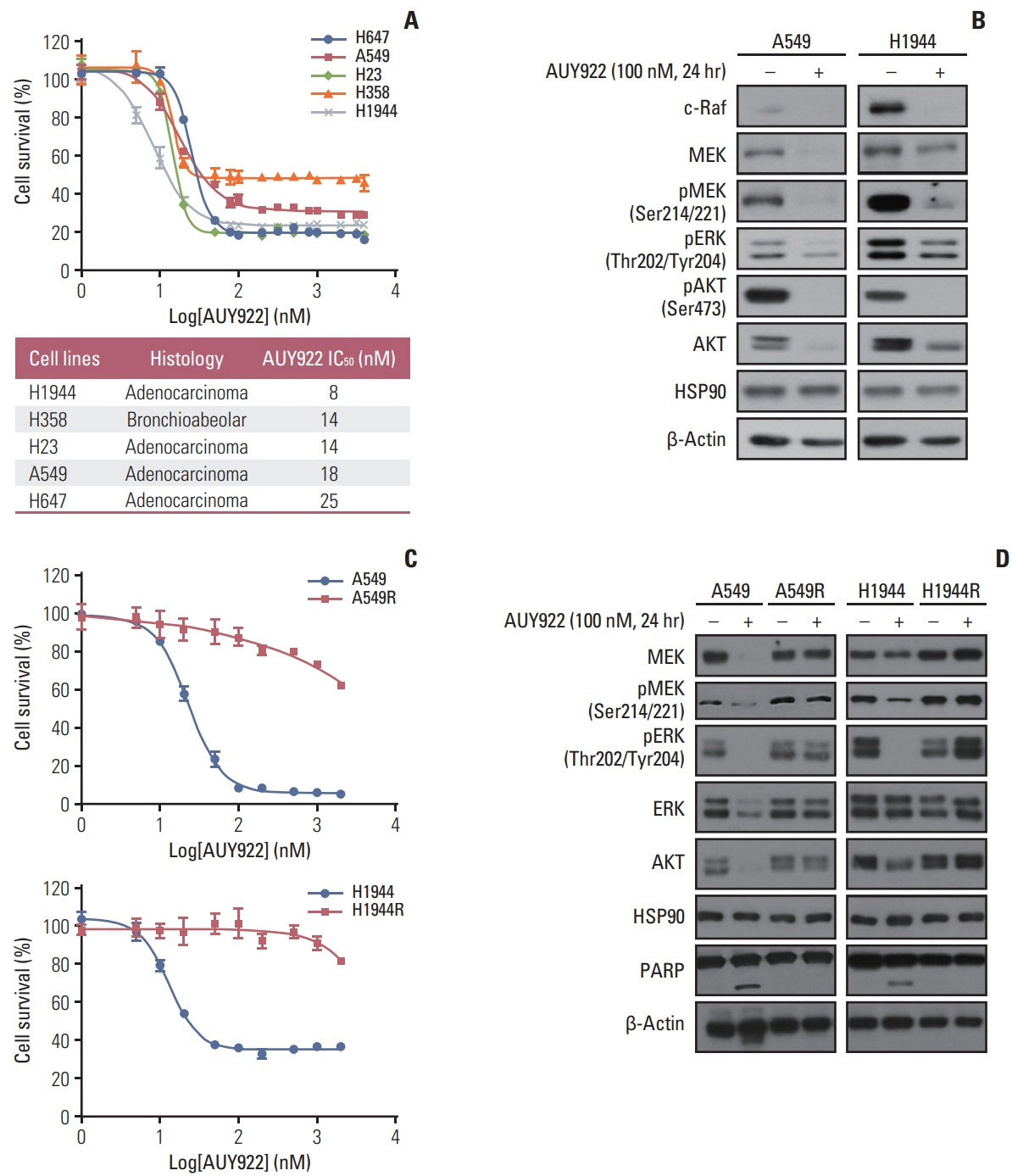
Fig. 1 Establishment of acquired AUY922 resistance in NSCLC cells harboring a KRAS mutation. (A) Antitumor effects of AUY922 alone against three KRAS-mutated NSCLC cell lines. NSCLC cells were treated with different concentrations of AUY922 for 72 hours. The results are expressed as a mean±SD of three independent experiments. (B) Results of western blotting analysis showing the endogenous protein levels in two KRAS-mutated NSCLC cell lines, A549 and H1944, after AUY922 treatment. (C) Anti-cancer effect of lapatinib in the A549 and H1944 cells, and their acquired AUY922-resistant counterparts, A549R and H1944R. Cell proliferation was measured using a CellTiter-Glo Luminescent Cell Viability Assay. The average results±SD of three independent experiments are shown. (D) Western blotting analysis of MEK, pMEK, pERK, ERK, AKT, HSP90, and PARP expression following incubation with increasing doses of AUY922 in the A549, A549R, H1944, and H1944R cell lines. β-Actin was included as a loading control (R, resistant cells). ERK, extracellular-signal-regulated kinase; HSP90, heat shock protein-90; MEK, mitogen-activated protein kinase; NSCLC, non–small cell lung cancer; PARP, poly(ADP-ribose) polymerase; SD, standard deviation.
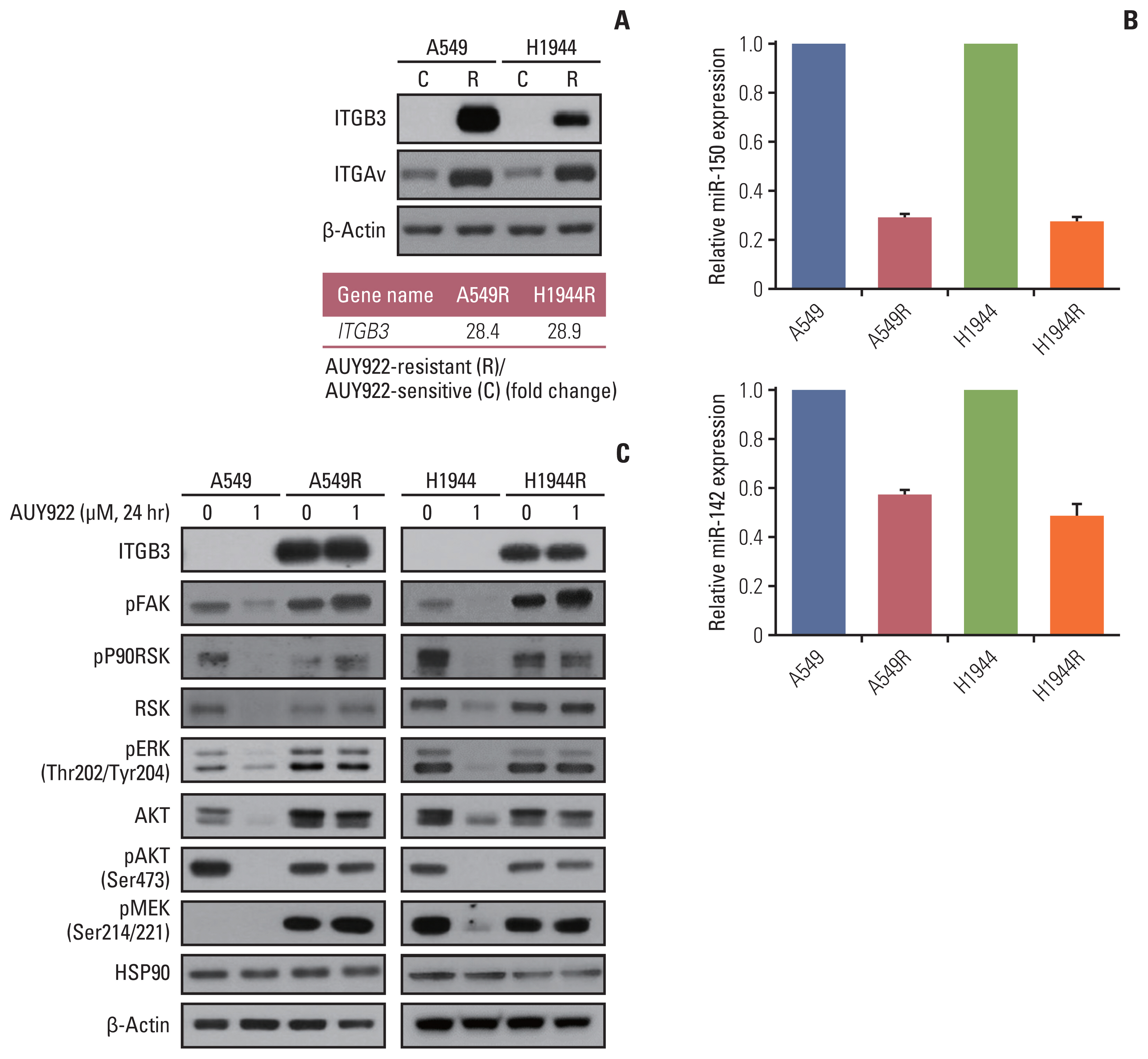
Fig. 2 FAK activation via ITGAvB3 induces AUY922 resistance in KRAS-mutated NSCLC cells. (A) Western blot analysis of the ITGB3 and ITGAv proteins in the acquired AUY922-resistant NSCLC cell lines (A549R and H1944R). (B) Real-time PCR analysis of miR-150 and miR-142 expression in the acquired AUY922-resistant NSCLC cell lines. (C) Western blotting analysis of ITGB3, pFAK, pRSK, RSK, pERK, AKT, pAKT, pMEK, and HSP90 expression in A549, A549R, H1944, and H1944R cell lines following incubation with 1 μM of AUY922. β-Actin was included as a loading control. FAK, focal adhesion kinase; ITGAv, integrin Av; ITGB3, integrin β3; NSCLC, non–small cell lung cancer; PCR, polymerase chain reaction.
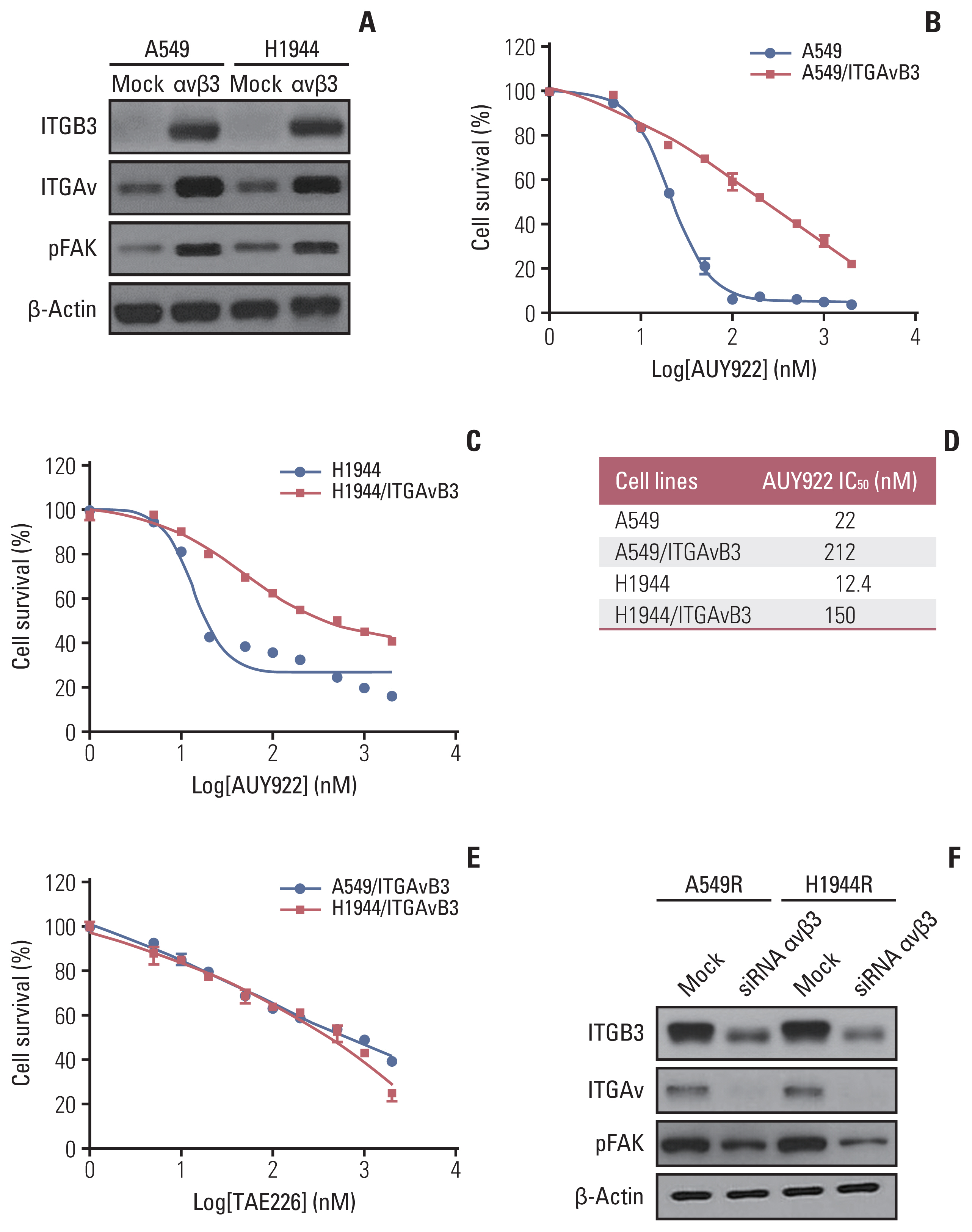
Fig. 3 Overexpression of ITGAv and ITGB3 induces AUY922 resistance in KRAS-mutated non–small cell lung cancer (NSCLC) cells. (A) Western blot analysis of ITGB3, ITGAv, and pFAK proteins in stably ITGAv and ITGB3 expressing NSCLC cell lines. (B, C) Effect of ITGAv and ITGB3 on AUY922 sensitivity of two KRAS-mutant NSCLC cell lines, A549/ITGAvB3 and H1944/ITGAvB3. CellTiter-Glo Luminescent Cell Viability Assays were performed 72 hours after drug treatment. Data represent the mean±SD of triplicate measurements relative to untreated cells. (D) IC50 values are reported in the table. (E) Anti-apoptotic effect of TAE226 in A549/ITGAvB3 and H1944/ITGAvB3 cell lines. CellTiter-Glo Luminescent Cell Viability Assays were performed 72 hours after drug treatment. (F) Western blot analysis of ITGB3, ITGAv, and pFAK proteins in stably ITGAv and ITGB3 knock-downed AUY922-resistant NSCLC cell lines. FAK, focal adhesion kinase; ITGAv, integrin Av; ITGB3, integrin β3; NSCLC, non–small cell lung cancer; SD, standard deviation.
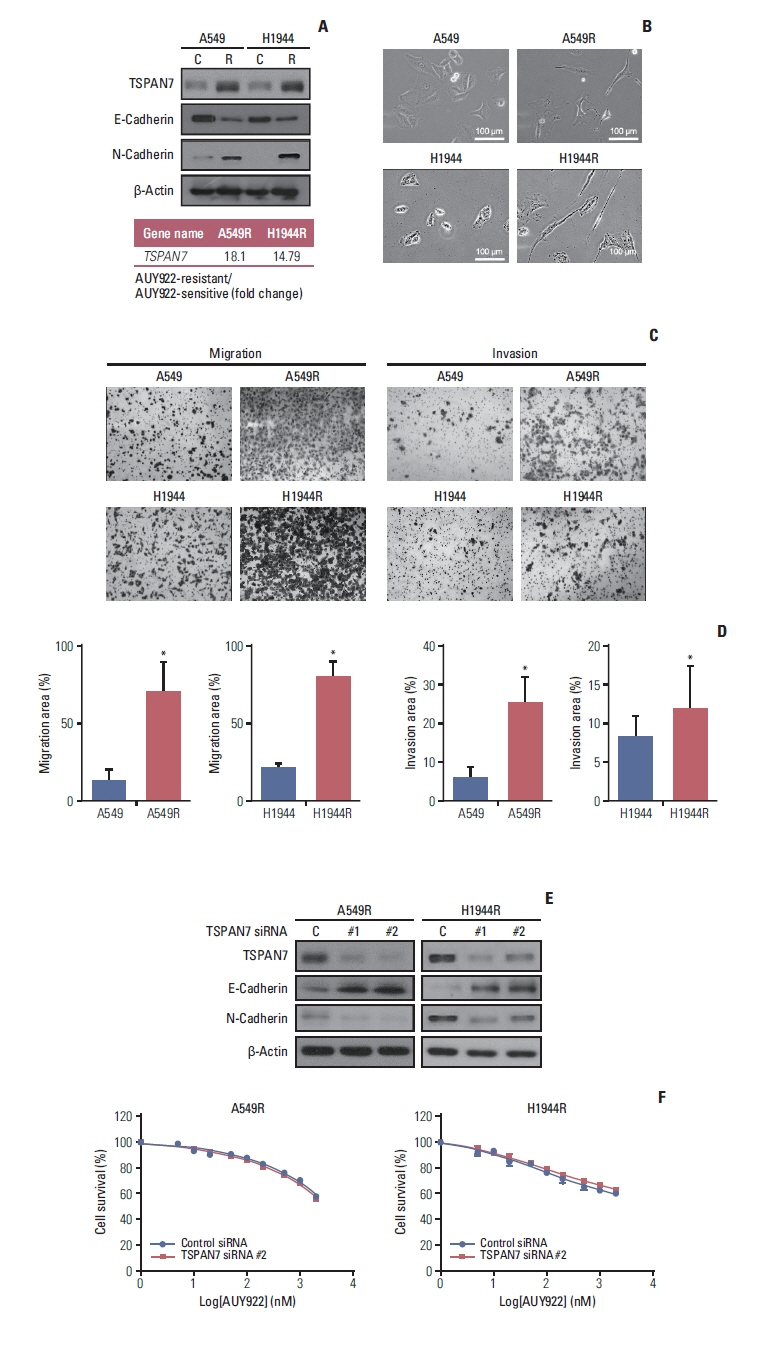
Fig. 4 TSPAN7 contributes to the epithelial-mesenchymal transition of acquired AUY922-resistant cells. (A) Western blot analysis of the epithelial-mesenchymal transition signaling proteins, TSPAN7, E-cadherin, and N-cadherin, in the two AUY922 resistant NSCLC cell lines, A549R and H1944R. (B) Phase-contrast images of the A549R and H1944R cells revealing notable morphological changes (×200). (C, D) Migration and invasion assay of the A549R and H1944R cells. These NSCLC cell lines were serum-starved for 24 hours and cell migration and invasion assays were conducted at 48 hours. Cell migration and invasion indexes were measured as described in the Methods. Data are representative of three independent experiments. *p < 0.05. (E) Western blot analysis of A549R and H1944R cells at 72 hours after TSPAN7 siRNA transfection. β-Actin was used as a loading control. (F) TSPAN7 siRNA could not rescue the AUY922 sensitivity in A549R and H1944R cells. Two days after transfection, two cell lines were treated with AUY922 for 3 days, followed by cell viability assay. The data represent triplicate experiments. NSCLC, non–small cell lung cancer; TSPAN, tetraspanin 7.
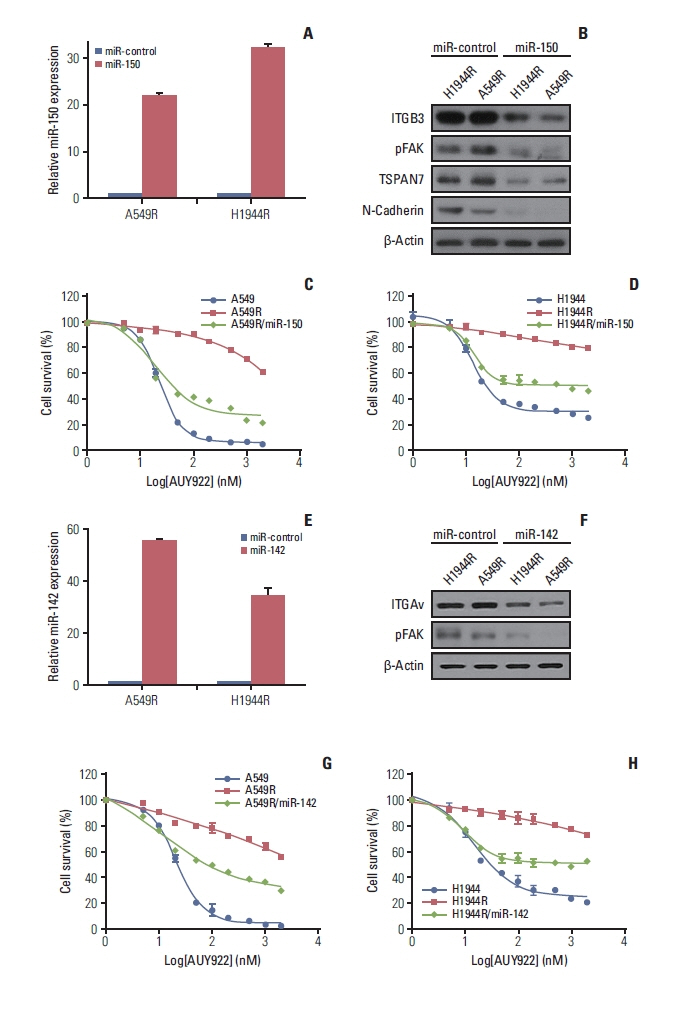
Fig. 5 FAK activation via miR-150 and miR-142 downregulation induce AUY922 resistance. (A) Real-time PCR analysis of ectopic miR-150 expression in A549R/miR-150 and H1944/miR-150 stable cells. (B) Effect of ectopic miR-150 expression on ITGB3, pFAK, TSPAN7 and N-cadherin in miR-control and miR-150 transfected resistant cells, revealed by western blot analysis. (C, D) Ectopic miR-150 expression renders A549R and H1944R cells sensitive to AUY922. Cell viability assays were performed at 72 hours after AUY922 treatment of the indicated cells at different concentrations. Data represent the mean±SD of triplicate experiments relative to untreated cells. (E) Real-time PCR analysis of ectopic miR-142 expression in A549R/miR-142 and H1944/miR-142 stable cells. (F) Effect of ectopic miR-142 expression on ITGB3, pFAK in miR-control and miR-142 transfected resistant cells, revealed by western blot analysis. (G, H) Ectopic miR-142 expression renders A549R and H1944R cells sensitive to AUY922. Cell viability assays were performed at 72 hours after AUY922 treatment of the indicated cells at different concentrations. Data represent the mean±SD of triplicate experiments relative to untreated cells. FAK, focal adhesion kinase; ITGAv, integrin Av; ITGB3, integrin β3; PCR, polymerase chain reaction; SD, standard deviation; TSPAN, tetraspanin 7.
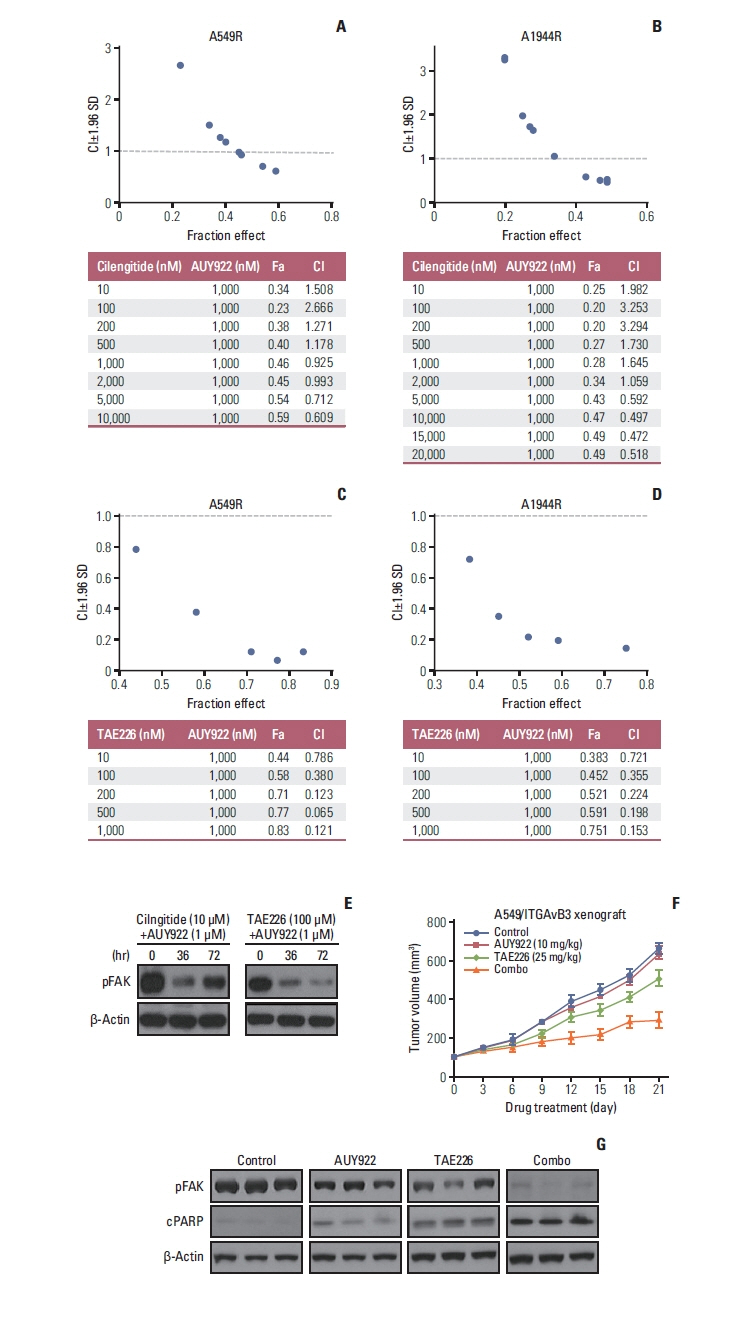
Fig. 6 A FAK inhibitor, and not an integrin inhibitor, shows synergistic effects with AUY922 in AUY922-resistant cells. (A, B) Synergistic effect of a cilengitide and AUY922 combination. Cell viability assays were performed in A549R and H1944R cells treated with 1,000 nM of AUY922 plus increasing concentrations of cilengitide for 3 days. (C, D) Synergistic effect of a TAE226 and AUY922 combination. Cell viability assays were performed in A549R and H1944R cells treated with 1,000 nM of AUY922 plus increasing concentrations of TAE226 for 3 days. The confidence interval (CI) was calculated using CalcuSyn software. (E) Western blot analysis of pFAK in A549R cells after each combination treatment. (F) Twelve mice harboring subcutaneous xenograft tumors derived from A549/ITGAvB3 cells were randomized into four groups. After the tumor volume reached 100 mm3, AUY922 at 10 mg/kg was administered 3 days/wk, and TAE226 (25 mg/kg) was administered 5 days/wk for up to 21 days. The tumor size was assessed at least three times a week. (G) Western blotting analysis detected cleaved PARP in the mouse xenograft tumors. CI, combi nation indexes; Fa, fraction affected; FAK, focal adhesion kinase; PARP, poly(ADP-ribose) polymerase; SD, standard deviation.
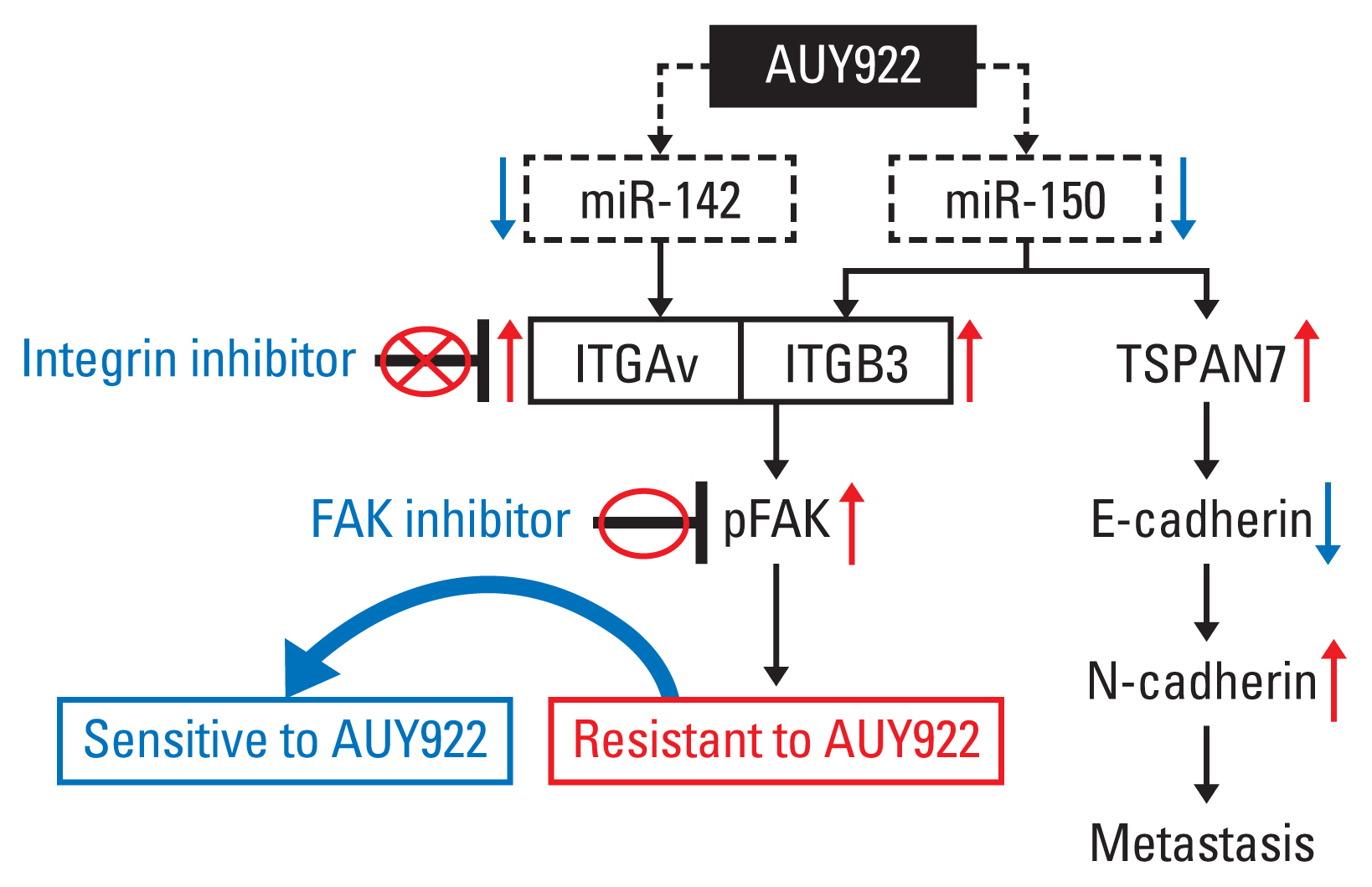
Fig. 7 Schemas illustrating the identified mechanisms of acquired resistance to AUY922. FAK, focal adhesion kinase; ITGAv, integrin Av; ITGB3, integrin β3; TSPAN, tetraspanin 7.
Reference
-
References
1. Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011; 11:775–91.
Article2. Park KS, Oh B, Lee MH, Nam KY, Jin HR, Yang H, et al. The HSP90 inhibitor, NVP-AUY922, sensitizes KRAS-mutant non-small cell lung cancer with intrinsic resistance to MEK inhibitor, trametinib. Cancer Lett. 2016; 372:75–81.
Article3. Park KS, Yang H, Choi J, Seo S, Kim D, Lee CH, et al. The HSP90 inhibitor, NVP-AUY922, attenuates intrinsic PI3K inhibitor resistance in KRAS-mutant non-small cell lung cancer. Cancer Lett. 2017; 406:47–53.
Article4. Papke B, Der CJ. Drugging RAS: know the enemy. Science. 2017; 355:1158–63.
Article5. Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016; 15:771–85.
Article6. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020; 383:1207–17.
Article7. Janne PA, Rybkin II, Spira AI, Riely GJ, Papadopoulos KP, Sabari JK, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/metastatic non-small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. Eur J Cancer. 2020; 138(Suppl 2):S1–2.8. Johnson ML, Ou SH, Barve M, Rybkin II, Papadopoulos KP, Leal TA, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with colorectal cancer (CRC) and other solid tumors harboring a KRAS G12C mutation. Eur J Cancer. 2020; 138(Suppl 2):S2.
Article9. Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol. 2013; 14:e358–69.
Article10. Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994; 91:8324–8.
Article11. Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, et al. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones. 1998; 3:100–8.
Article12. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005; 5:761–72.
Article13. Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, Picard D. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones. 2018; 23:467–82.
Article14. Sidera K, Patsavoudi E. HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov. 2014; 9:1–20.
Article15. Seggewiss-Bernhardt R, Bargou RC, Goh YT, Stewart AK, Spencer A, Alegre A, et al. Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer. 2015; 121:2185–92.
Article16. Ambros V. The functions of animal microRNAs. Nature. 2004; 431:350–5.
Article17. Ma J, Dong C, Ji C. MicroRNA and drug resistance. Cancer Gene Ther. 2010; 17:523–31.
Article18. Schwickert A, Weghake E, Bruggemann K, Engbers A, Brinkmann BF, Kemper B, et al. microRNA miR-142-3p inhibits breast cancer cell invasiveness by synchronous targeting of WASL, integrin alpha V, and additional cytoskeletal elements. PLoS One. 2015; 10:e0143993.
Article19. Borschel CS, Stejskalova A, Schafer SD, Kiesel L, Gotte M. miR-142-3p reduces the size, migration, and contractility of endometrial and endometriotic stromal cells by targeting integrin- and Rho GTPase-related pathways that regulate cytoskeletal function. Biomedicines. 2020; 8:291.
Article20. Honda N, Jinnin M, Kira-Etoh T, Makino K, Kajihara I, Makino T, et al. miR-150 down-regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin beta3. Am J Pathol. 2013; 182:206–16.21. Guan JL. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life. 2010; 62:268–76.
Article22. Alanko J, Ivaska J. Endosomes: emerging platforms for integrin-mediated FAK signalling. Trends Cell Biol. 2016; 26:391–8.
Article23. Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006; 18:516–23.
Article24. Wang X, Lin M, Zhao J, Zhu S, Xu M, Zhou X. TSPAN7 promotes the migration and proliferation of lung cancer cells via epithelial-to-mesenchymal transition. Onco Targets Ther. 2018; 11:8815–22.
Article25. Chen W, Harbeck MC, Zhang W, Jacobson JR. MicroRNA regulation of integrins. Transl Res. 2013; 162:133–43.
Article26. Busacca S, Law EW, Powley IR, Proia DA, Sequeira M, Le Quesne J, et al. Resistance to HSP90 inhibition involving loss of MCL1 addiction. Oncogene. 2016; 35:1483–92.
Article27. Mumin NH, Drobnitzky N, Patel A, Lourenco LM, Cahill FF, Jiang Y, et al. Overcoming acquired resistance to HSP90 inhibition by targeting JAK-STAT signalling in triple-negative breast cancer. BMC Cancer. 2019; 19:102.
Article28. Kumar CC. Integrin alpha v beta 3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets. 2003; 4:123–31.29. Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004; 5:816–26.
Article30. Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev Cell. 2008; 14:843–53.
Article31. Suetsugu T, Koshizuka K, Seki N, Mizuno K, Okato A, Arai T, et al. Downregulation of matrix metalloproteinase 14 by the antitumor miRNA, miR-150-5p, inhibits the aggressiveness of lung squamous cell carcinoma cells. Int J Oncol. 2018; 52:913–24.
Article32. Zhang Z, Wang J, Li J, Wang X, Song W. MicroRNA-150 promotes cell proliferation, migration, and invasion of cervical cancer through targeting PDCD4. Biomed Pharmacother. 2018; 97:511–7.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- CD98 activation increases surface expression and clusteringof beta 1 integrins in MCF-7 cells through FAK/Src- and cytoskeleton-independent mechanisms
- ATP-induced focal adhesion kinase activity is negatively modulated by phospholipase D2 in PC12 cells
- SUV39H1 is a New Client Protein of Hsp90 Degradated by Chaetocin as a Novel C-Terminal Inhibitor of Hsp90
- Role of Integrin, FAK (Focal Adhesion Kinase) and ERK (Extracellular Signal Regulated Kinase) on the Suppressed Cell Proliferation of Endometrial Cancer Cells by GnRH (Gonadotropin-Releasing Hormone)
- Mechanisms of Acquired Resistance to Epidermal Growth Factor Receptor Inhibitors and Overcoming Strategies in Lung Cancer
