Two Compound Heterozygous Were Identified in SLC26A4 Gene in Two Chinese Families With Enlarged Vestibular Aqueduct
- Affiliations
-
- 1Beijing Key Laboratory for Pediatric Diseases of Otolaryngology, Head and Neck Surgery, MOE Key Laboratory of Major Diseases in Children, Beijing Pediatric Research Institute, National Center for Children's Health, Beijing, China. nixin@bch.com.cn guoyongli@bch.com.cn
- 2Department of Otolaryngology, Head and Neck Surgery, Beijing Children's Hospital, Capital Medical University, National Center for Children's Health, Beijing, China.
- KMID: 2437491
- DOI: http://doi.org/10.21053/ceo.2018.00213
Abstract
OBJECTIVES
To investigate the genetic causes of hearing loss with enlarged vestibular aqueduct (EVA) in two children from unrelated two Chinese families.
METHODS
Sanger sequencing of all coding exons in SLC26A4 (encoding Pendrin protein) was performed on the two patients, their sibling and parents respectively. To predict and visualize the potential functional outcome of the novel variant, model building, structure analysis, and in silico analysis were further conducted.
RESULTS
The results showed that the proband from family I harbored a compound heterozygote of SLC26A4 c.1174A>T (p.N392Y) mutation and c.1181delTCT (p.F394del) variant in exon 10, potentially altering Pendrin protein structure. In family II, the proband was identified in compound heterozygosity with a known mutation of c.919-2A>G in the splice site of intron 7 and a novel mutation of c.1023insC in exon 9, which results in a frameshift and translational termination, consequently leading to truncated Pendrin protein. Sequence homology analysis indicated that all the mutations localized at high conservation sites, which emphasized the significance of these mutations on Pendrin spatial organization and function.
CONCLUSION
In summary, this study revealed two compound heterozygous mutations (c.1174A>T/c.1181delTCT; c.919- 2A>G/c.1023insC) in Pendrin protein, which might account for the deafness of the two probands clinically diagnosed with EVA. Thus this study contributes to improve understanding of the causes of hearing loss associated with EVA and develop a more scientific screening strategy for deafness.
MeSH Terms
Figure
-

Fig. 1. Clinical phenotype presentations of the two hearing loss children. (A) Bilateral play audiometry detection. The X-axis indicates frequency in hertz (Hz) and the Y-axis indicates hearing level in decibels (dB HL). (B) The temporal bone computed tomography scan of the two probands shows the bilateral enlarged vestibular aqueduct (arrows).
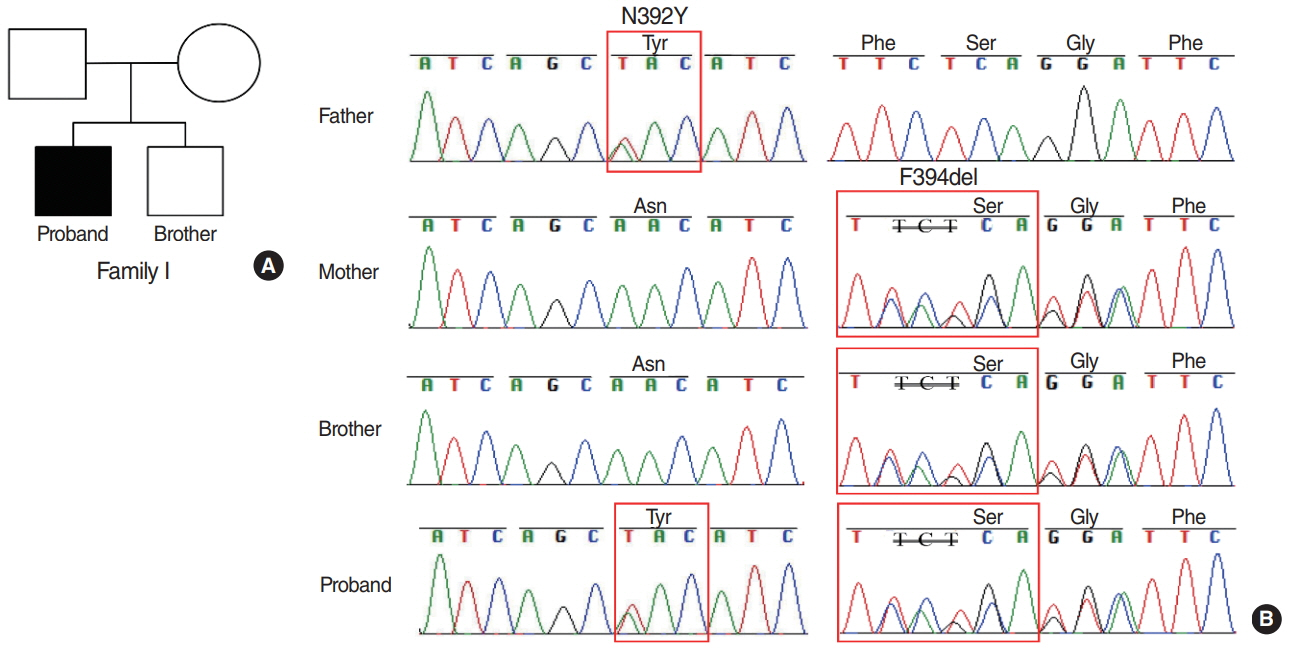
Fig. 2. Pedigree map and sequence electropherograms in family I. (A) Pedigree map. Squares and circles denote males and females, respectively. (B) Sequence electropherograms showed wild type or abnormal sequence from four members of family I. The red box shows site of the heterozygous mutation of c.1174A>T and c.1181delTCT in the SLC26A4 gene.

Fig. 3. Protein alignment showed conservation of p.N392 and p.F394 residues across 10 species. These two mutations occurred at evolutionarily conserved amino acids (red arrows). The red helixes mark α-helix regions in protein structure.

Fig. 4. Illustration of the three-dimensional structure of the wild-type and mutant Pendrin protein. (A) Wide type. (B) p.N392Y. (C) p.F394del.
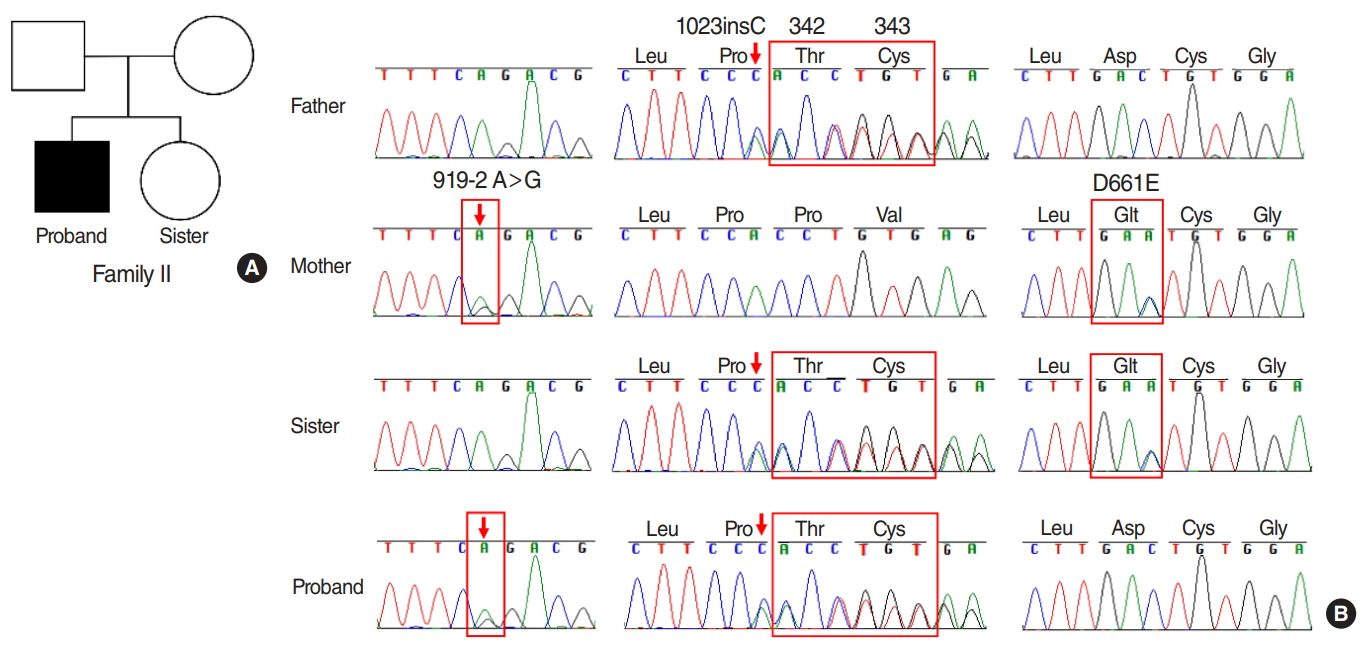
Fig. 5. Pedigree map and sequence electropherograms in family II. (A) Pedigree map. Squares and circles denote males and females, respectively. (B) Sequence electropherograms showed wild type or abnormal sequence from four members of family II. The red box shows site of the novel heterozygous mutation of c.919-2A>G, c.1023insC and c.1983C>A in the SLC26A4 gene.

Fig. 6. Wide type and mutants of Pendrin alignment in family II. (A) Protein alignment showed that the novel mutation of c.1023insC induced a frameshift mutation, caused a stop codon at position of 376 amino acid, resulting in truncated of Pendrin protein. (B) The putative schematic representation of Pendrin protein and mutants in family II. Notes: both c.919-2A>G and p.D661E were known deleterious mutations.
Reference
-
1. Morton CC, Nance WE. Newborn hearing screening: a silent revolution. N Engl J Med. 2006; May. 354(20):2151–64.2. Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull. 2002; 63:73–94.
Article3. Lin YH, Lin YH, Lu YC, Liu TC, Chen CY, Hsu CJ, et al. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci Rep. 2017; Aug. 7(1):7551.
Article4. Korver AM, Smith RJ, Van Camp G, Schleiss MR, Bitner-Glindzicz MA, Lustig LR, et al. Congenital hearing loss. Nat Rev Dis Primers. 2017; Jan. 3:16094.
Article5. Du W, Wang Q, Zhu Y, Wang Y, Guo Y. Associations between GJB2, mitochondrial 12S rRNA, SLC26A4 mutations, and hearing loss among three ethnicities. Biomed Res Int. 2014; 2014:746838.
Article6. Fang Y, Gu M, Wang C, Suo F, Wang G, Xia Y. GJB2 as well as SLC26A4 gene mutations are prominent causes for congenital deafness. Cell Biochem Biophys. 2015; Sep. 73(1):41–4.
Article7. Han S, Yang X, Zhou Y, Hao J, Shen A, Xu F, et al. Deafness gene mutations in newborns in Beijing. Acta Otolaryngol. 2016; 136(5):475–9.
Article8. Jiang Y, Huang S, Deng T, Wu L, Chen J, Kang D, et al. Mutation spectrum of common deafness-causing genes in patients with non-syndromic deafness in the Xiamen Area, China. PLoS One. 2015; Aug. 10(8):e0135088.
Article9. Ladsous M, Vlaeminck-Guillem V, Dumur V, Vincent C, Dubrulle F, Dhaenens CM, et al. Analysis of the thyroid phenotype in 42 patients with Pendred syndrome and nonsyndromic enlargement of the vestibular aqueduct. Thyroid. 2014; Apr. 24(4):639–48.
Article10. Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011; May. 32(5):557–63.
Article11. Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H, Zong L, et al. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet. 2007; Sep. 72(3):245–54.
Article12. Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, et al. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006; Jun. 14(6):773–9.
Article13. Shearer AE, Hildebrand MS, Smith RJ. Hereditary hearing loss and deafness overview. In : Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, editors. GeneReview. Seattle (WA): University of Washington;1993-2018.14. Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978; May. 88(5):723–8.
Article15. Zhang F, Bai X, Xiao Y, Zhang X, Zhang G, Li J, et al. Identification of a novel mutation in SLC26A4 gene in a Chinese family with enlarged vestibular aqueduct syndrome. Int J Pediatr Otorhinolaryngol. 2016; Jun. 85:75–9.
Article16. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. Swiss-model: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014; Jul. 42(Web Server issue):W252–8.
Article17. Fu C, Zheng H, Zhang S, Chen Y, Su J, Wang J, et al. Mutation screening of the SLC26A4 gene in a cohort of 192 Chinese patients with congenital hypothyroidism. Arch Endocrinol Metab. 2016; Aug. 60(4):323–7.
Article18. Zhao FF, Lan L, Wang DY, Han B, Qi Y, Zhao Y, et al. Correlation analysis of genotypes, auditory function, and vestibular size in Chinese children with enlarged vestibular aqueduct syndrome. Acta Otolaryngol. 2013; Dec. 133(12):1242–9.
Article19. Chan DK, Chang KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope. 2014; Feb. 124(2):E34–53.
Article20. Zhang J, Wang P, Han B, Ding Y, Pan L, Zou J, et al. Newborn hearing concurrent genetic screening for hearing impairment: a clinical practice in 58,397 neonates in Tianjin, China. Int J Pediatr Otorhinolaryngol. 2013; Dec. 77(12):1929–35.21. Anwar S, Riazuddin S, Ahmed ZM, Tasneem S, Khan SY, et al. SLC26A4 mutation spectrum associated with DFNB4 deafness and Pendred’s syndrome in Pakistanis. J Hum Genet. 2009; May. 54(5):266–70.
Article22. Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, et al. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001; May. 17(5):403–11.23. Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S, Usami S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet. 2003; Dec. 11(12):916–22.
Article24. Xin F, Yuan Y, Deng X, Han M, Wang G, Zhao J, et al. Genetic mutations in nonsyndromic deafness patients of Chinese minority and Han ethnicities in Yunnan, China. J Transl Med. 2013; Dec. 11:312.
Article25. Li Q, Zhu QW, Yuan YY, Huang SS, Han DY, Huang DL, et al. Identification of SLC26A4 c.919-2A>G compound heterozygosity in hearing-impaired patients to improve genetic counseling. J Transl Med. 2012; Nov. 10:225.
Article26. Yong AM, Goh SS, Zhao Y, Eng PH, Koh LK, Khoo DH. Two Chinese families with Pendred’s syndrome: radiological imaging of the ear and molecular analysis of the pendrin gene. J Clin Endocrinol Metab. 2001; Aug. 86(8):3907–11.27. Ishihara K, Okuyama S, Kumano S, Iida K, Hamana H, Murakoshi M, et al. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear Res. 2010; Dec. 270(1-2):110–8.
Article28. Sharma AK, Rigby AC, Alper SL. STAS domain structure and function. Cell Physiol Biochem. 2011; 28(3):407–22.
Article29. Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, et al. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. 2004; Apr. 6(4):343–50.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Family of H723R Mutation for SLC26A4 Associated with Enlarged Vestibular Aqueduct Syndrome
- A Novel Frameshift Mutation of SLC26A4 in a Korean Family With Nonsyndromic Hearing Loss and Enlarged Vestibular Aqueduct
- Compound Heterozygosity for Two Novel SLC26A4 Mutations in a Large Iranian Pedigree with Pendred Syndrome
- Familial Enlarged Vestibular Aqueduct Syndrome (FEVAS)
- Two Cases of Enlarged Vestibular Aqueduct Syndrome
