Non-Type I Cystinuria Associated with Mental retardation and Ataxia in a Korean Boy with a New Missence Mutation(G173R) in the SLC7A9 Gene
- Affiliations
-
- 1Department of Pediatrics and Research Laboratory for Mitochondrial Disorders, Ajou University School of Medicine, Suwon, Korea. pedkim@ajou.ac.kr
- KMID: 1713853
- DOI: http://doi.org/10.3346/jkms.2010.25.1.172
Abstract
- Cystinuria is an inherited renal and intestinal disease characterized by defective amino acids reabsorption and cystine urolithiasis. It is unusually associated with neurologic symptoms. Mutations in two genes, SLC3A1 and SLC7A9, have been identified in cystinuric patients. This report presents a 13-yr-old boy with cystinuria who manifested difficulty in walking, ataxia, and mental retardation. Somatosensory evoked potential of posterior tibial nerve stimulation showed the central conduction dysfunction through the posterior column of spinal cord. He was diagnosed non-type I cystinuria by urinary amino acid analysis and oral cystine loading test. We screened him and his family for gene mutation by direct sequencing of SLC3A1 and SLC7A9 genes. In this patient, we identified new missence mutation G173R in SLC7A9 gene.
MeSH Terms
-
Adolescent
Amino Acid Substitution
Amino Acid Transport Systems, Basic/*genetics
Amino Acids/urine
Ataxia/complications/diagnosis/*genetics
Base Sequence
Cystine/blood
Cystinuria/complications/diagnosis/*genetics
Humans
Intellectual Disability/complications/diagnosis/*genetics
Male
*Mutation, Missense
Pedigree
Republic of Korea
Amino Acid Transport Systems, Basic
Amino Acids
Cystine
Figure
-
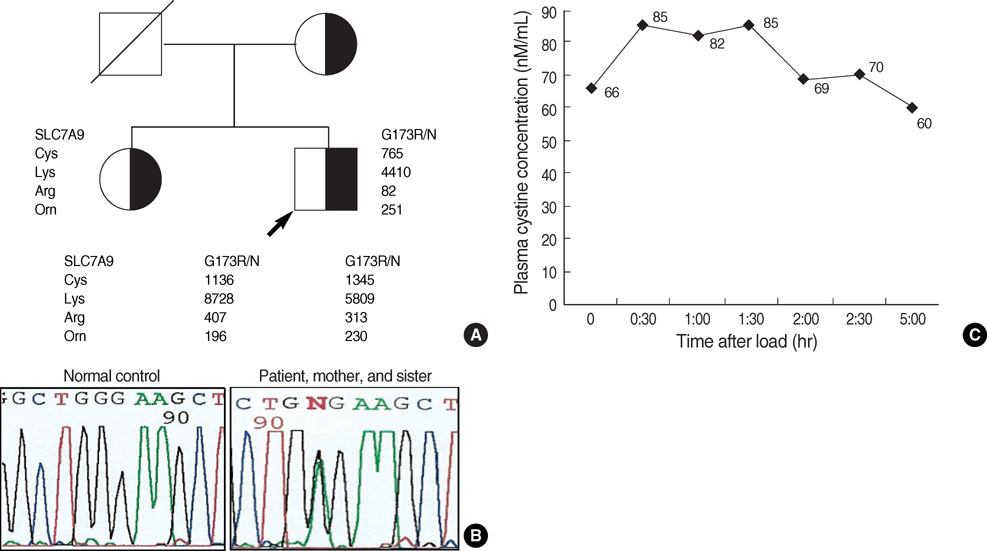
Fig. 1 Pedigree of family with patient that carry mutation in SLC7A9, electropherograms of DNA sequencing for G173R mutation, and the result of oral cystine load in the patient. (A) Pedigree of family with patient; mutation in SLC7A9 and urine dibasic amino acid excretion. Urine amino acid excretion values for cystine (Cys), lysine (Lys), arginine (Arg), and ornithine (Orn) are expressed in µM/g creatinine (normal range: Cys, 30-130; Lys, 150-630; Arg, 30-110; Orn, 30-90). (B) Point mutation G173R (535G→A substitution) in SLC7A9 gene localized in exon 5. Patient's sequence has both G (normal) and A (mutant) at nucleotide 535, resulting in the presence of Gly (codon GGA) and Arg (codon AGA). (C) Changes in plasma cystine level after an oral L-cystine dihydrochloride load of 0.25 mM/kg body weight in the patient.
Reference
-
1. Font-Llitjos M, Jimenez-Vidal M, Bisceglia L, Di Perna M, de Sanctis L, Rousaud F, Zelante L, Palacin M, Nunes V. New insights into cystinuria: 40 new mutations, genotype-phenotype correlation, and digenic inheritance causing partial phenotype. J Med Genet. 2005. 42:58–68.
Article2. Calonge MJ, Gasparini P, Chillaron J, Chillon M, Galluci M, Rousaud F, Zelante L, Testar X, Dallapiccola B, Di Silverio F, Barcelo P, Estivill X, Zorano A, Nunes V, Palacin M. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet. 1994. 6:420–425.
Article3. Feliubadalo L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, Golomb E, Centola M, Aksentijevich I, Kreiss Y, Goldman B, Pras M, Kastner DM, Pras E, Gasparini P, Bisceglia L, Beccia E, Galluci M, de Sanctis L, Ponzone A, Rizzoni GF, Zelante L, Bassi MT, George AL, Palacin M. Non-type I cystinuria caused by nutations in SLC7A9, encording a subunit(bo, +AT) of rBAT. International Cystinuria Consortium. Nat Genet. 1999. 23:52–57.4. Scriver CR, Whelan DT, Clow CL, Dallaire L. Cystinuria: increased prevalence in patients with mental disease. N Engl J Med. 1970. 283:783–786.
Article5. De Myer W, Gebhard RL. Subacute combined degeneration of the spinal cord with cystinuria. Neurology. 1975. 25:994–997.
Article6. Cavanagh NP, Bicknell J, Howard F. Cystinuria with mental retardation and paroxysmal dyskinesia in 2 brothers. Arch Dis Child. 1974. 49:662–664.
Article7. Font MA, Feliubadalo L, Estivill X, Nunes V, Golomb E, Kreiss Y, Pras E, Bisceglia L, d'Adamo AP, Zelante L, Gasparini P, Bassi MT, George AL Jr, Manzoni M, Riboni M, Ballabio A, Borsani G, Reig N, Fernandez E, Zorzano A, Bertran J, Palacin M. International Cystinuria Consortium. Functional analysis of mutations in SLC7A9, and genotype-phenotype correlation in non-Type I cystinuria. Hum Mol Genet. 2001. 10:305–316.
Article8. Milliner DS. Cystinuria. Endocrinol Metab Clin North Am. 1990. 19:889–907.
Article9. Bannai S. Transport of cystine and cysteine in mammalian cells. Biochim Biophys Acta. 1984. 779:289–306.
Article10. Hwang SM, Weiss S, Segal S. Uptake of L-[35S]cystine by isolated rat brain capillaries. J Neurochem. 1980. 35:417–424.11. Guillen M, Corella D, Cabello ML, Garcia AM, Hernandez-Yago J. Reference values of urinary excretion of cystine and dibasic amino acids: classification of patients with cystinuria in the Valencian Community, Spain. Clin Biochem. 1999. 32:25–30.12. Jaeken J, Martens K, Francois I, Eyskens F, Lecointre C, Derua R, Meulemans S, Slootstra JW, Waelkens E, de Zegher F, Creemers JW, Matthijs G. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am J Hum Genet. 2006. 78:38–51.
Article13. Parvari R, Brodyansky I, Elpeleg O, Moses S, Landau D, Hershkovitz E. A recessive contiguous gene deletion of chromosome 2p16 associated with cystinuria and a mitochondrial disease. Am J Hum Genet. 2001. 69:869–875.
Article14. Chabrol B, Martens K, Meulemans S, Cano A, Jaeken J, Matthijs G, Creemers JW. Deletion of C2orf34, PREPL and SLC3A1 causes atypical hypotonia-cystinuria syndrome. J Med Genet. 2008. 45:314–318.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Genotype and Phenotype Analysis in Pediatric Patients with Cystinuria
- The First Case of X-linked Alpha-thalassemia/Mental Retardation (ATR-X) Syndrome in Korea
- A case of cystinuria with a heterozygous SLC3A1 mutation presenting with recurrent multiple renal stones in a 14-year-old boy
- Episodic Ataxia Type 2 due to a Deletion Mutation in the CACNA1A Gene in a Korean Family
- A Patient Diagnosed with Spinocerebellar Ataxia Type 5 associated with SPTBN2: Case Report
