Glucolipotoxicity Suppressed Autophagy and Insulin Contents in Human Islets, and Attenuation of PERK Activity Enhanced Them in an ATG7-Dependent Manner
- Affiliations
-
- 1Department of Internal Medicine, Seoul National University Hospital, Seoul, Korea
- 2Department of Surgery and Cancer Research Institute, Seoul National University Hospital, Seoul, Korea
- KMID: 2553594
- DOI: http://doi.org/10.4093/dmj.2022.0366
Abstract
- Background
Administration of pancreatic endoplasmic reticulum kinase inhibitor (PERKi) improved insulin secretion and hyperglycemia in obese diabetic mice. In this study, autophagic balance was studied whether to mediate it.
Methods
Human islets were isolated from living patients without diabetes. PERKi GSK2606414 effects were evaluated in the islets under glucolipotoxicity by palmitate. Islet insulin contents and secretion were measured. Autophagic flux was assessed by microtubule associated protein 1 light chain 3 (LC3) conversion, a red fluorescent protein (RFP)-green fluorescent protein (GFP)- LC3 tandem assay, and P62 levels. For mechanical analyses, autophagy was suppressed using 3-methyladenine in mouse islets. Small interfering RNA for an autophagy-related gene autophagy related 7 (Atg7) was transfected to interfere autophagy.
Results
PERKi administration to mice decreased diabetes-induced P62 levels in the islets. Glucolipotoxicity significantly increased PERK phosphorylation by 70% and decreased insulin contents by 50% in human islets, and addition of PERKi (40 to 80 nM) recovered both. PERKi also enhanced glucose-stimulated insulin secretion (6-fold). PERKi up-regulated LC3 conversion suppressed by glucolipotoxicity, and down-regulated P62 contents without changes in P62 transcription, indicating enhanced autophagic flux. Increased autophagosome-lysosome fusion by PERKi was visualized in mouse islets, where PERKi enhanced ATG7 bound to LC3. Suppression of Atg7 eliminated PERKi-induced insulin contents and secretion.
Conclusion
This study provided functional changes of human islets with regard to autophagy under glucolipotoxicity, and suggested modulation of autophagy as an anti-diabetic mechanism of PERKi.
Keyword
Figure
-

Fig. 1. Low-dose pancreatic endoplasmic reticulum kinase inhibitor (PERKi) treatment decreased P62 levels in the islets of obese diabetic mice. PERKi GSK2656157 (10 mg/kg/day) was administered for 8 weeks to diabetic mice induced by a high-fat diet and streptozotocin injections, in accordance with the Institutional Animal Care and Use Committee of Seoul National University Hospital (SNU-150327-3-2), as previously described [10]. After euthanasia, pancreas was harvested and preserved in paraffin blocks. Section slides were stained for insulin and P62 in brown, and quantification of the P62 intensity per islet area was calculated. Animal numbers 5–6/group and 10.1 ± 1.8 islets were analyzed in each mouse. (A) immunohistochemical staining for P62 and insulin and (B) P62 staining intensity. (A) Representative images with islets delineated by dashed lines. (B) The lines and error bars on the plot represent mean ± standard error of the mean. One-way analysis of variance (ANOVA) was applied. DM, diabetes mellitus; C, control; V, vehicle.
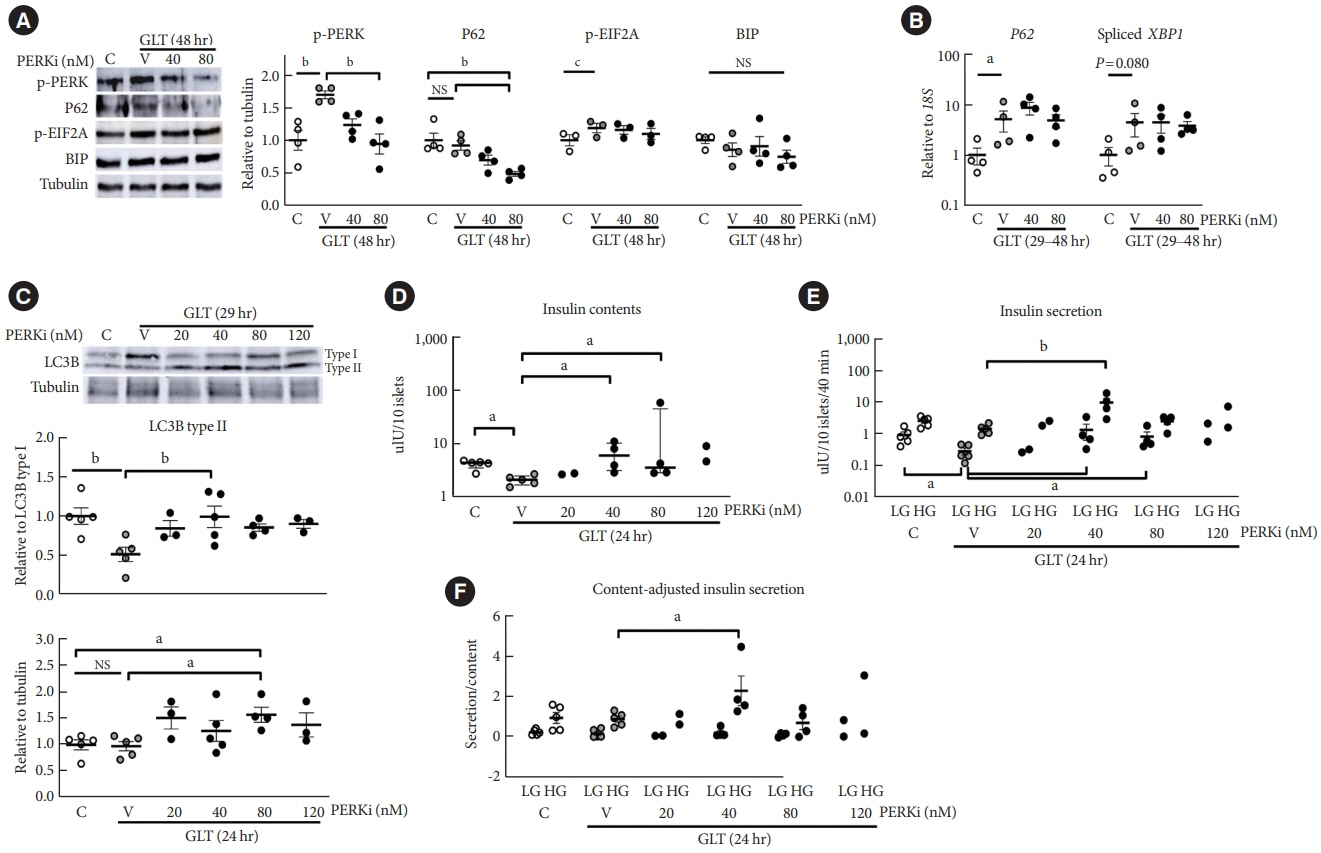
Fig. 2. Low-dose pancreatic endoplasmic reticulum kinase inhibitor (PERKi) enhanced autophagy activity, insulin contents, and insulin secretion in human islets under chronic glucolipotoxicity (GLT). Human islets were isolated from living donors’ pancreas and exposed to GLT (glucose 20 mM and palmitate 0.5 mM) for 29 to 48 hours, with/without GSK2606414. Each treatment group was composed of 25 to 30 islets, resulting in total 120 islets per experiment, by hand-picking of healthy islets estimated by their morphology. The islets were randomly assigned to control, vehicle, and PERKi groups, without blinding to the researchers. (A, C) Western blots and (B) quantitative reverse transcription-polymerase chain reaction of autophagy and unfolded protein response markers. In (C), bafilomycin A (4 nM) was added to the culture medium during the last 5 hours. (D) Islet insulin content, (E) insulin secretion, and (F) insulin secretion adjusted by the islet insulin contents. Because of large inter-participant variation, (B, D, E) are shown on a log-10 scale. In (A, C), one-way analysis of variance (ANOVA) with Bonferroni posttest was used, except for phospho-eukaryotic translation initiation factor 2 alpha (EIF2A) where paired t-test was applied due to limited sample size. For (B), unpaired t-test was used between the control and vehicle. For (D), the Kruskal-Wallis with Dunn’s multiple comparison test was applied. In (E, F), two-way repeated measures ANOVA with Bonferroni posttest was applied. For (B, D, E), logarithmic transformation was performed before the analyses. Experiment numbers 4 to 5. The lines and error bars represent median (interquartile ranges) for (D) and mean ± standard error of the mean (all the others). C, control; V, vehicle; BIP, binding immunoglobulin protein; NS, no significant differences; LC3B, light chain 3B; LG, low glucose; HG, high glucose. aP<0.05 and bP<0.01 between the groups, cP < 0.01 in the paired t-test.

Fig. 3. Low-dose pancreatic endoplasmic reticulum kinase inhibitor (PERKi) improved 3-methyladenine (3-MA)-suppressed autophagy and insulin synthesis through autophagy related 7 (ATG7). (A-E) Mouse islets or dispersed cells were treated with 3-MA (0.5 mM) for 24 hours with/without GSK2606414. Autophagy was induced by fetal bovine serum deprivation by further incubation for 5 hours (A, B). Each treatment group was composed of 25 to 30 islets, resulting in total 120 islets from 3 to 4 mice per experiment, by hand-picking of healthy islets estimated by their morphology. The islets were randomly assigned to the groups, without blinding to the researchers. Western blotting of autophagy and unfolded protein response markers was conducted, and representative blots are demonstrated (A, C, D, E). For (B), dispersed islet cells were transduced with BacMam tandem red fluorescent protein (RFP)-green fluorescent protein (GFP)-light chain 3B (LC3B) reagent, incubated overnight, then treated as described above. Before fixation with 4% formaldehyde, live cell nuclei were stained with Hoechst 33342, and GFP or RFP puncta were manually counted in cytoplasm of live cells. About 100 cells were counted in each treatment group by 3 to 6 experiments. Representative pictures are presented: yellow puncta indicating quenching of acid-sensitive GFP by lysosome (in the control), a green punctate (3-MA group) and red puncta indicating autophagolysosome (3-MA with PERKi group) marked by arrows. (F, G, H) Mouse islets were dispersed to single cells, and transfected with 100 nM of siRNA for Atg7 and negative control. Then autophagy was inhibited by 3-MA (0.5 mM) with/without GSK2606414 (20 nM). Each treatment group was composed of 105 cells, resulting in total 3 × 105 cells from 5 to 6 mice per experiment. The cells were randomly assigned to the groups, without blinding to the researchers. After 30 to 48 hours, quantitative reverse transcription-polymerase chain reaction was conducted (Atg7 and insulin 1 [Ins1]), and protein levels were measured by Western blot (ATG7 and tubulin) and enzyme-linked immunosorbent assay (ELISA; insulin contents and secretion). Dot numbers in each group represent experimental numbers. The lines and error bars are median (interquartile ranges) (E) and mean ± standard error of the mean (all the others). One-way analysis of variance (ANOVA) with Bonferroni posttest was applied, except for (B, E) where Kruskal-Wallis with Dunn's multiple comparison test was applied excluding the serum-free control in (E), and for (H) where two-way repeated measures ANOVA with Dunnett's multiple comparisons test was applied. C, control; V, vehicle; NS, no significant differences; EIF2A, eukaryotic translation initiation factor 2 alpha; BIP, binding immunoglobulin protein; LG, low glucose; HG, high glucose; siAtg7, Atg7 siRNA. aP < 0.05, bP < 0.01 in the post hoc analyses.
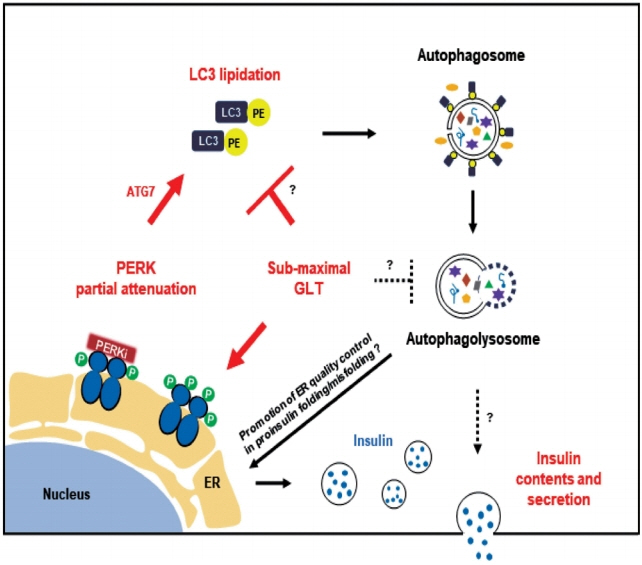
Fig. 4. A working hypothesis of pancreatic endoplasmic reticulum kinase inhibitor (PERKi) action in human β-cells under glucolipotoxicity (GLT). Metabolic stress suppresses microtubule associated protein 1 light chain 3 (LC3) lipidation and low-dose PERKi recovers it, causing enhanced autophagic activity and subsequent improvement in endoplasmic reticulum (ER) function. Especially, ER-phagy would enhance clearance of misfolded proinsulin, which contributes to efficient proinsulin translation/post-translational modification [38]. Observations in this study is shown in red, and inference from literature in black. PE, phosphatidylethanolamine; ATG7, autophagy related 7; P, phosphorylation.
Reference
-
1. Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002; 110:1389–98.
Article2. Kefalas G, Larose L. PERK leads a hub dictating pancreatic b cell homoeostasis. Biol Cell. 2018; 110:27–32.3. Moon S, Jung HS. Endoplasmic reticulum stress and dysregulated autophagy in human pancreatic beta cells. Diabetes Metab J. 2022; 46:533–42.
Article4. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl] acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012; 55:7193–207.5. Wang R, McGrath BC, Kopp RF, Roe MW, Tang X, Chen G, et al. Insulin secretion and Ca2+ dynamics in b-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J Biol Chem. 2013; 288:33824–36.6. Harding HP, Zyryanova AF, Ron D. Uncoupling proteostasis and development in vitro with a small molecule inhibitor of the pancreatic endoplasmic reticulum kinase, PERK. J Biol Chem. 2012; 287:44338–44.
Article7. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013; 73:1993–2002.
Article8. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013; 5:206ra138.
Article9. Kim MJ, Min SH, Shin SY, Kim MN, Lee H, Jang JY, et al. Attenuation of PERK enhances glucose-stimulated insulin secretion in islets. J Endocrinol. 2018; 236:125–36.
Article10. Kim MJ, Kim MN, Min SH, Ham DS, Kim JW, Yoon KH, et al. Specific PERK inhibitors enhanced glucose-stimulated insulin secretion in a mouse model of type 2 diabetes. Metabolism. 2019; 97:87–91.
Article11. Jung HS, Chung KW, Kim JW, Kim J, Komatsu M, Tanaka K, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008; 8:318–24.12. Kim J, Park K, Kim MJ, Lim H, Kim KH, Kim SW, et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat Commun. 2021; 12:183.
Article13. Marasco MR, Linnemann AK. B-cell autophagy in diabetes pathogenesis. Endocrinology. 2018; 159:2127–41.14. Yoon JW, Jung HS, Jang JY, Kim MJ, Kim JH, Ohn JH, et al. Improved insulin secretion by autologous islet transplantation, compared to oral antidiabetic agents, after distal pancreatectomy. Cell Transplant. 2015; 24:1615–26.
Article15. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016; 12:1–222.16. Marselli L, Piron A, Suleiman M, Colli ML, Yi X, Khamis A, et al. Persistent or transient human b cell dysfunction induced by metabolic stress: specific signatures and shared gene expression with type 2 diabetes. Cell Rep. 2020; 33:108466.17. Lytrivi M, Castell AL, Poitout V, Cnop M. Recent insights into mechanisms of b-cell lipo- and glucolipotoxicity in type 2 diabetes. J Mol Biol. 2020; 432:1514–34.18. Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000; 408:488–92.
Article19. Lee YH, Kim J, Park K, Lee MS. B-cell autophagy: mechanism and role in b-cell dysfunction. Mol Metab. 2019; 27S(Suppl):S92–103.20. Frudd K, Burgoyne T, Burgoyne JR. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun. 2018; 9:95.
Article21. Hart NJ, Powers AC. Use of human islets to understand islet biology and diabetes: progress, challenges and suggestions. Diabetologia. 2019; 62:212–22.
Article22. Barovic M, Distler M, Schoniger E, Radisch N, Aust D, Weitz J, et al. Metabolically phenotyped pancreatectomized patients as living donors for the study of islets in health and diabetes. Mol Metab. 2019; 27S(Suppl):S1–6.
Article23. Vivot K, Pasquier A, Goginashvili A, Ricci R. Breaking bad and breaking good: b-cell autophagy pathways in diabetes. J Mol Biol. 2020; 432:1494–513.24. Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, Masini M, et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes. 2014; 63:1978–93.
Article25. Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia. 2009; 52:1083–6.
Article26. Martino L, Masini M, Novelli M, Beffy P, Bugliani M, Marselli L, et al. Palmitate activates autophagy in INS-1E b-cells and in isolated rat and human pancreatic islets. PLoS One. 2012; 7:e36188.27. Zummo FP, Krishnanda SI, Georgiou M, O’Harte FP, Parthsarathy V, Cullen KS, et al. Exendin-4 stimulates autophagy in pancreatic b-cells via the RAPGEF/EPAC-Ca2+-PPP3/calcineurin-TFEB axis. Autophagy. 2022; 18:799–815.
Article28. Mir SU, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE. Inhibition of autophagic turnover in b-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem. 2015; 290:6071–85.29. Trudeau KM, Colby AH, Zeng J, Las G, Feng JH, Grinstaff MW, et al. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J Cell Biol. 2016; 214:25–34.
Article30. Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in b-cells. J Biol Chem. 2011; 286:42534–44.31. Zummo FP, Cullen KS, Honkanen-Scott M, Shaw JA, Lovat PE, Arden C. Glucagon-like peptide 1 protects pancreatic bcells from death by increasing autophagic flux and restoring lysosomal function. Diabetes. 2017; 66:1272–85.32. Roomp K, Kristinsson H, Schvartz D, Ubhayasekera K, Sargsyan E, Manukyan L, et al. Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PLoS One. 2017; 12:e0176391.
Article33. Weir GC. Glucolipotoxicity, b-cells, and diabetes: the emperor has no clothes. Diabetes. 2020; 69:273–8.34. Yagishita Y, Fukutomi T, Sugawara A, Kawamura H, Takahashi T, Pi J, et al. Nrf2 protects pancreatic b-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes. 2014; 63:605–18.35. Allen D, Seo J. ER stress activates the TOR pathway through Atf6. J Mol Signal. 2018; 13:1.
Article36. Blackwood EA, Hofmann C, Santo Domingo M, Bilal AS, Sarakki A, Stauffer W, et al. ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, Rheb. Circ Res. 2019; 124:79–93.
Article37. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019; 176:11–42.
Article38. Liu M, Huang Y, Xu X, Li X, Alam M, Arunagiri A, et al. Normal and defective pathways in biogenesis and maintenance of the insulin storage pool. J Clin Invest. 2021; 131:e142240.
Article39. Uchizono Y, Alarcon C, Wicksteed BL, Marsh BJ, Rhodes CJ. The balance between proinsulin biosynthesis and insulin secretion: where can imbalance lead? Diabetes Obes Metab. 2007; 9 Suppl 2:56–66.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Mitochondrial Complexes I and II Are More Susceptible to Autophagy Deficiency in Mouse beta-Cells
- Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic beta-cells
- Vitrification, in vitro fertilization, and development of Atg7 deficient mouse oocytes
- Repopulation of autophagy-deficient stromal cells with autophagy-intact cells after repeated breeding in uterine mesenchyme-specific Atg7 knockout mice
- Partial Deletion of Perk Improved High-Fat Diet-Induced Glucose Intolerance in Mice
