The Puzzling Case of Hyperexcitability in Amyotrophic Lateral Sclerosis
- Affiliations
-
- 1Department of Neurology, College of Medicine, Inje University, Busan, Korea.
- 2Neuroscience Research Australia, Sydney, Australia. M.kiernan@unsw.edu.au
- 3Department of Neurology, Westmead Clinical School, University of Sydney, Westmead, Australia.
- 4Multidisciplinary Motor Neurone Disease Clinical Service, Prince of Wales Clinical School, University of New South Wales, Sydney, Australia.
- KMID: 2179162
- DOI: http://doi.org/10.3988/jcn.2013.9.2.65
Abstract
- The development of hyperexcitability in amyotrophic lateral sclerosis (ALS) is a well-known phenomenon. Despite controversy as to the underlying mechanisms, cortical hyperexcitability appears to be closely related to the interplay between excitatory corticomotoneurons and inhibitory interneurons. Hyperexcitability is not a static phenomenon but rather shows a pattern of progression in a spatiotemporal aspect. Cortical hyperexcitability may serve as a trigger to the development of anterior horn cell degeneration through a 'dying forward' process. Hyperexcitability appears to develop during the early disease stages and gradually disappears in the advanced stages of the disease, linked to the destruction of corticomotorneuronal pathways. As such, a more precise interpretation of these unique processes may provide new insight regarding the pathophysiology of ALS and its clinical features. Recently developed technologies such as threshold tracking transcranial magnetic stimulation and automated nerve excitability tests have provided some clues about underlying pathophysiological processes linked to hyperexcitability. Additionally, these novel techniques have enabled clinicians to use the specific finding of hyperexcitability as a useful diagnostic biomarker, enabling clarification of various ALS-mimic syndromes, and the prediction of disease development in pre-symptomatic carriers of familial ALS. In terms of nerve excitability tests for peripheral nerves, an increase in persistent Na+ conductances has been identified as a major determinant of peripheral hyperexcitability in ALS, inversely correlated with the survival in ALS. As such, the present Review will focus primarily on the puzzling theory of hyperexcitability in ALS and summarize clinical and pathophysiological implications for current and future ALS research.
Keyword
MeSH Terms
Figure
-
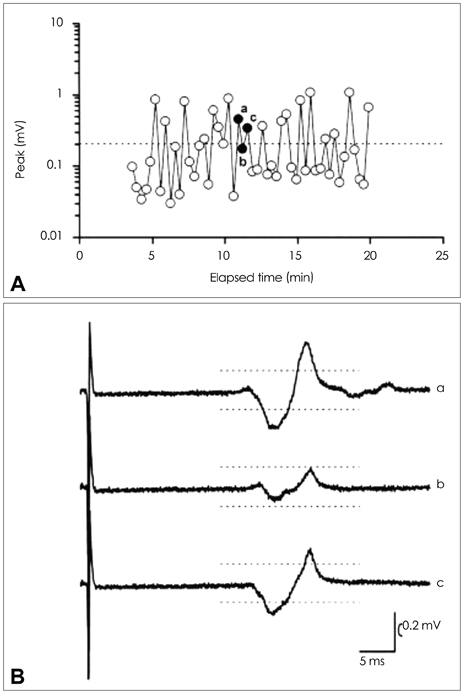
Fig. 1 Threshold tracking transcranial magnetic stimulation (TT-TMS) protocol to identify the cortical excitability.52 The dashed horizontal line represents the target output of 0.2 mV (peak to peak) which was "tracked". The circles (clear and filled) represent the magnitude of the motor evoked potential (MEP) amplitude with each stimulus (A). Illustrated three MEP responses of different amplitude track are tracked. The MEP response is initially larger (a), then smaller (b), and again larger (c) than the target output of 0.2 mV in three consecutive stimuli (B) (permission from John Wiley and Sons).
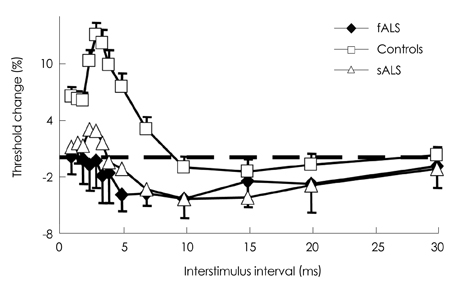
Fig. 2 Short interval intracortical inhibition in fALS and sALS. SICI is decreased in patients who are clinically affected with fALS, who express a mutation in SOD1, and in patients with sALS in comparison to healthy controls. Modified from Vucic et al.51 (permission from Oxford University Press). fALS: familial amyotrophic lateral sclerosis, sALS: sporadic amyotrophic lateral sclerosis, SICI: short-inhibitory cortical interval, SOD1: superoxide dismutase type 1.
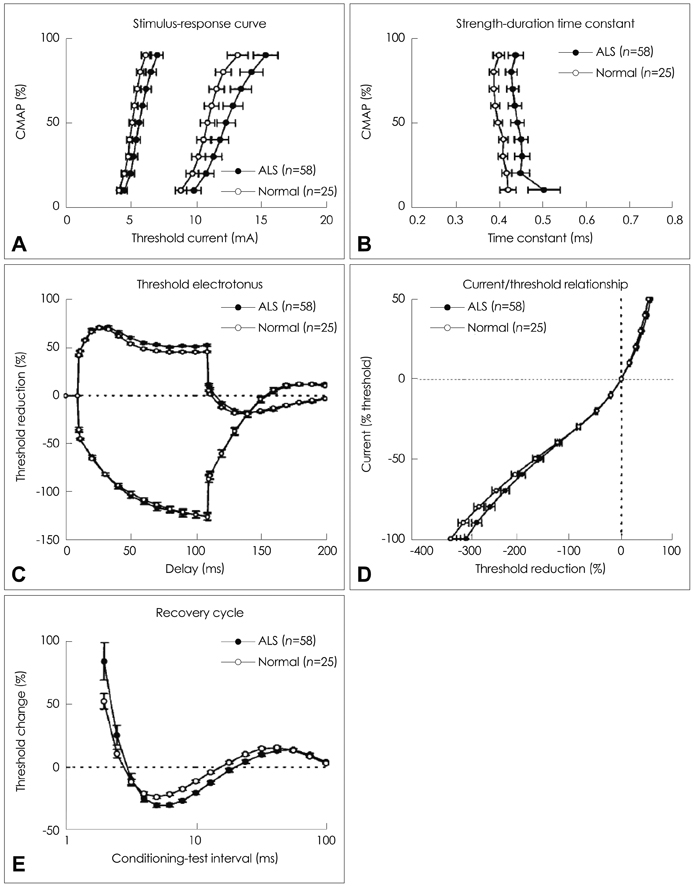
Fig. 3 Grand-averaged data of multiple excitability measurements in patients with ALS (n=58; filled circle) and in age-matched normal subjects (n=25; open circle).69 In Stimulus-Response Curve, left curves were obtained by stimulus duration=1.0 ms; right curves were obtained by stimulus duration=0.2 ms. Each plot denotes various parameters of axonal excitability: stimulation-response curve (A), strength-duration time constant (B), threshold electrotonus (C), current/threshold relationship (D), recovery cycle (E). Error bars indicate SEM (permission from Oxford University Press). ALS: amyotrophic lateral sclerosis, SEM: standard error mean.
Cited by 1 articles
-
Dissociation of Structural and Functional Integrities of the Motor System in Amyotrophic Lateral Sclerosis and Behavioral-Variant Frontotemporal Dementia
Jong Seok Bae, Michele Ferguson, Rachel Tan, Eneida Mioshi, Neil Simon, James Burrell, Steve Vucic, John R. Hodges, Matthew C Kiernan, Michael Hornberger
J Clin Neurol. 2016;12(2):209-217. doi: 10.3988/jcn.2016.12.2.209.
Reference
-
1. Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011. 377:942–955.
Article2. Kiernan MC. Hyperexcitability, persistent Na+ conductances and neurodegeneration in amyotrophic lateral sclerosis. Exp Neurol. 2009. 218:1–4.
Article3. Section I: Alphabetic list of terms with definitions. Muscle Nerve. 2001. 24:S5–S28.4. Kleine BU, Stegeman DF, Schelhaas HJ, Zwarts MJ. Firing pattern of fasciculations in ALS: evidence for axonal and neuronal origin. Neurology. 2008. 70:353–359.
Article5. Swash M. Why are upper motor neuron signs difficult to elicit in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2012. 83:659–662.
Article6. Scheel AK, Toepfer M, Kunkel M, Finkenstaedt M, Reimers CD. Ultrasonographic assessment of the prevalence of fasciculations in lesions of the peripheral nervous system. J Neuroimaging. 1997. 7:23–27.
Article7. Van der Heijden A, Spaans F, Reulen J. Fasciculation potentials in foot and leg muscles of healthy young adults. Electroencephalogr Clin Neurophysiol. 1994. 93:163–168.
Article8. Mills KR. Motor neuron disease. Studies of the corticospinal excitation of single motor neurons by magnetic brain stimulation. Brain. 1995. 118:971–982.9. de Carvalho M, Miranda PC, Lourdes Sales Luís M, Ducla-Soares E. Neurophysiological features of fasciculation potentials evoked by transcranial magnetic stimulation in amyotrophic lateral sclerosis. J Neurol. 2000. 247:189–194.
Article10. Mills KR, Nithi KA. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve. 1997. 20:1137–1141.
Article11. Eisen A. Amyotrophic lateral sclerosis-Evolutionary and other perspectives. Muscle Nerve. 2009. 40:297–304.
Article12. de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008. 119:497–503.
Article13. Turner MR, Kiernan MC. Does interneuronal dysfunction contribute to neurodegeneration in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. 2012. 13:245–250.
Article14. Mazzini L, Balzarini C, Gareri F, Brigatti M. H-reflex changes in the course of amyotrophic lateral sclerosis. Electroencephalogr Clin Neurophysiol. 1997. 104:411–417.
Article15. Raynor EM, Shefner JM. Recurrent inhibition is decreased in patients with amyotrophic lateral sclerosis. Neurology. 1994. 44:2148–2153.
Article16. de Carvalho M, Swash M. Cramps, muscle pain, and fasciculations: not always benign? Neurology. 2004. 63:721–723.
Article17. de Carvalho M. Why is ALS so excited? Clin Neurophysiol. 2011. 122:1689–1690.
Article18. Krarup C. Lower motor neuron involvement examined by quantitative electromyography in amyotrophic lateral sclerosis. Clin Neurophysiol. 2011. 122:414–422.
Article19. Hallett M. Transcranial magnetic stimulation and the human brain. Nature. 2000. 406:147–150.
Article20. Hallett M. Transcranial magnetic stimulation: a primer. Neuron. 2007. 55:187–199.
Article21. Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve. 1998. 21:137–158.
Article22. Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009. 73:805–811.
Article23. Chou SM, Norris FH. Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve. 1993. 16:864–869.
Article24. Eisen A, Kim S, Pant B. Amyotrophic lateral sclerosis (ALS): a phylogenetic disease of the corticomotoneuron? Muscle Nerve. 1992. 15:219–224.
Article25. Maragakis NJ, Rothstein JD. Glutamate transporters in neurologic disease. Arch Neurol. 2001. 58:365–370.
Article26. Zeman S, Lloyd C, Meldrum B, Leigh PN. Excitatory amino acids, free radicals and the pathogenesis of motor neuron disease. Neuropathol Appl Neurobiol. 1994. 20:219–231.
Article27. Rothstein JD. Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis. Adv Neurol. 1995. 68:7–20. discussion 21-27.28. Watkins JC, Evans RH. Excitatory amino acid transmitters. Annu Rev Pharmacol Toxicol. 1981. 21:165–204.
Article29. Heath PR, Shaw PJ. Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve. 2002. 26:438–458.
Article30. Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990. 11:379–387.
Article31. Regan RF, Panter SS, Witz A, Tilly JL, Giffard RG. Ultrastructure of excitotoxic neuronal death in murine cortical culture. Brain Res. 1995. 705:188–198.
Article32. Maher P, Davis JB. The role of monoamine metabolism in oxidative glutamate toxicity. J Neurosci. 1996. 16:6394–6401.
Article33. Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, et al. On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006. 8:2075–2087.
Article34. Druga R. Neocortical inhibitory system. Folia Biol (Praha). 2009. 55:201–217.35. Ling LL, Hughes LF, Caspary DM. Age-related loss of the GABA synthetic enzyme glutamic acid decarboxylase in rat primary auditory cortex. Neuroscience. 2005. 132:1103–1113.
Article36. Hestrin S, Galarreta M. Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 2005. 28:304–309.
Article37. Schwartz RD, Yu X, Katzman MR, Hayden-Hixson DM, Perry JM. Diazepam, given postischemia, protects selectively vulnerable neurons in the rat hippocampus and striatum. J Neurosci. 1995. 15:529–539.
Article38. Chen Q, Moulder K, Tenkova T, Hardy K, Olney JW, Romano C. Excitotoxic cell death dependent on inhibitory receptor activation. Exp Neurol. 1999. 160:215–225.
Article39. Eisen A, Pant B, Stewart H. Cortical excitability in amyotrophic lateral sclerosis: a clue to pathogenesis. Can J Neurol Sci. 1993. 20:11–16.
Article40. Nihei K, McKee AC, Kowall NW. Patterns of neuronal degeneration in the motor cortex of amyotrophic lateral sclerosis patients. Acta Neuropathol. 1993. 86:55–64.
Article41. Lloyd CM, Richardson MP, Brooks DJ, Al-Chalabi A, Leigh PN. Extramotor involvement in ALS: PET studies with the GABA(A) ligand [(11)C]flumazenil. Brain. 2000. 123:2289–2296.
Article42. Foerster BR, Callaghan BC, Petrou M, Edden RA, Chenevert TL, Feldman EL. Decreased motor cortex gamma-aminobutyric acid in amyotrophic lateral sclerosis. Neurology. 2012. 78:1596–1600.
Article43. Petri S, Krampfl K, Hashemi F, Grothe C, Hori A, Dengler R, et al. Distribution of GABAA receptor mRNA in the motor cortex of ALS patients. J Neuropathol Exp Neurol. 2003. 62:1041–1051.
Article44. Baloh RH. How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr Opin Neurol. 2012. 25:701–707.
Article45. Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain. 2006. 129:2436–2446.
Article46. de Carvalho M, Pinto S, Swash M. Does the motor cortex influence denervation in ALS? EMG studies of muscles with both contralateral and bilateral corticospinal innervation. Clin Neurophysiol. 2011. 122:629–635.
Article47. Swash M, Ingram D. Preclinical and subclinical events in motor neuron disease. J Neurol Neurosurg Psychiatry. 1988. 51:165–168.
Article48. de Carvalho M, Swash M. The onset of ALS? Clin Neurophysiol. 2010. 121:1709–1710.
Article49. Rowland LP. Diagnosis of amyotrophic lateral sclerosis. J Neurol Sci. 1998. 160:Suppl 1. S6–S24.
Article50. Swash M. Early diagnosis of ALS/MND. J Neurol Sci. 1998. 160:Suppl 1. S33–S36.
Article51. Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008. 131:1540–1550.
Article52. Vucic S, Howells J, Trevillion L, Kiernan MC. Assessment of cortical excitability using threshold tracking techniques. Muscle Nerve. 2006. 33:477–486.
Article53. Yokota T, Yoshino A, Inaba A, Saito Y. Double cortical stimulation in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1996. 61:596–600.
Article54. Ziemann U, Winter M, Reimers CD, Reimers K, Tergau F, Paulus W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis. Evidence from paired transcranial magnetic stimulation. Neurology. 1997. 49:1292–1298.
Article55. Stefan K, Kunesch E, Benecke R, Classen J. Effects of riluzole on cortical excitability in patients with amyotrophic lateral sclerosis. Ann Neurol. 2001. 49:536–539.
Article56. Ziemann U. TMS and drugs. Clin Neurophysiol. 2004. 115:1717–1729.
Article57. Caramia MD, Palmieri MG, Desiato MT, Iani C, Scalise A, Telera S, et al. Pharmacologic reversal of cortical hyperexcitability in patients with ALS. Neurology. 2000. 54:58–64.
Article58. Eisen A. Clinical electrophysiology of the upper and lower motor neuron in amyotrophic lateral sclerosis. Semin Neurol. 2001. 21:141–154.
Article59. Chen R, Lozano AM, Ashby P. Mechanism of the silent period following transcranial magnetic stimulation. Evidence from epidural recordings. Exp Brain Res. 1999. 128:539–542.
Article60. Werhahn KJ, Kunesch E, Noachtar S, Benecke R, Classen J. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J Physiol. 1999. 517:591–597.
Article61. Drory VE, Kovach I, Groozman GB. Electrophysiologic evaluation of upper motor neuron involvement in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001. 2:147–152.
Article62. Zanette G, Tamburin S, Manganotti P, Refatti N, Forgione A, Rizzuto N. Changes in motor cortex inhibition over time in patients with amyotrophic lateral sclerosis. J Neurol. 2002. 249:1723–1728.
Article63. Turner MR, Hammers A, Al-Chalabi A, Shaw CE, Andersen PM, Brooks DJ, et al. Distinct cerebral lesions in sporadic and 'D90A' SOD1 ALS: studies with [11C]flumazenil PET. Brain. 2005. 128:1323–1329.
Article64. Vucic S, Kiernan MC. Cortical excitability testing distinguishes Kennedy's disease from amyotrophic lateral sclerosis. Clin Neurophysiol. 2008. 119:1088–1096.
Article65. Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007. 78:849–852.
Article66. Zanette G, Tamburin S, Manganotti P, Refatti N, Forgione A, Rizzuto N. Different mechanisms contribute to motor cortex hyperexcitability in amyotrophic lateral sclerosis. Clin Neurophysiol. 2002. 113:1688–1697.
Article67. Kanai K, Shibuya K, Sato Y, Misawa S, Nasu S, Sekiguchi Y, et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012. 83:734–738.
Article68. Mogyoros I, Kiernan MC, Burke D, Bostock H. Strength-duration properties of sensory and motor axons in amyotrophic lateral sclerosis. Brain. 1998. 121:851–859.
Article69. Kanai K, Kuwabara S, Misawa S, Tamura N, Ogawara K, Nakata M, et al. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain. 2006. 129:953–962.
Article70. Nakata M, Kuwabara S, Kanai K, Misawa S, Tamura N, Sawai S, et al. Distal excitability changes in motor axons in amyotrophic lateral sclerosis. Clin Neurophysiol. 2006. 117:1444–1448.
Article71. Vucic S, Kiernan MC. Axonal excitability properties in amyotrophic lateral sclerosis. Clin Neurophysiol. 2006. 117:1458–1466.
Article72. Vucic S, Krishnan AV, Kiernan MC. Fatigue and activity dependent changes in axonal excitability in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007. 78:1202–1208.
Article73. Cheah BC, Lin CS, Park SB, Vucic S, Krishnan AV, Kiernan MC. Progressive axonal dysfunction and clinical impairment in amyotrophic lateral sclerosis. Clin Neurophysiol. 2012. 123:2460–2467.
Article74. Stys PK, Waxman SG, Ransom BR. Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol. 1991. 30:375–380.
Article75. Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol. 2009. 215:368–379.
Article76. Eisen A, Kuwabara S. The split hand syndrome in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012. 83:399–403.
Article77. Wilbourn AJ. The "split hand syndrome". Muscle Nerve. 2000. 23:138.
Article78. Kuwabara S, Sonoo M, Komori T, Shimizu T, Hirashima F, Inaba A, et al. Dissociated small hand muscle atrophy in amyotrophic lateral sclerosis: frequency, extent, and specificity. Muscle Nerve. 2008. 37:426–430.
Article79. Voermans NC, Schelhaas HJ, Munneke M, Zwarts MJ. Dissociated small hand muscle atrophy in aging: the 'senile hand' is a split hand. Eur J Neurol. 2006. 13:1381–1384.
Article80. Kuwabara S, Mizobuchi K, Ogawara K, Hattori T. Dissociated small hand muscle involvement in amyotrophic lateral sclerosis detected by motor unit number estimates. Muscle Nerve. 1999. 22:870–873.
Article81. Bae JS, Sawai S, Misawa S, Kanai K, Isose S, Kuwabara S. Differences in excitability properties of FDI and ADM motor axons. Muscle Nerve. 2009. 39:350–354.
Article82. Menon P, Bae JS, Mioshi E, Kiernan MC, Vucic S. Split-hand plus sign in ALS: Differential involvement of the flexor pollicis longus and intrinsic hand muscles. Amyotroph Lateral Scler Frontotemporal Degener. Forthcoming 2012.
Article83. Clark CM, Forman MS. Frontotemporal lobar degeneration with motor neuron disease: a clinical and pathological spectrum. Arch Neurol. 2006. 63:489–490.
Article84. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006. 314:130–133.
Article85. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006. 351:602–611.
Article86. Forman MS, Trojanowski JQ, Lee VM. TDP-43: a novel neurodegenerative proteinopathy. Curr Opin Neurobiol. 2007. 17:548–555.
Article87. Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008. 63:535–538.88. Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008. 319:1668–1672.
Article89. Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009. 136:1001–1004.
Article90. Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009. 323:1208–1211.
Article91. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009. 323:1205–1208.
Article92. Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011. 134:2582–2594.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Syndrome of Progressive Bulbar Palsy in Amyotrophic Lateral Sclerosis: A Case Report
- Amyotrophic Lateral Sclerosis Associated With CADASIL
- Apraxia of Eyelid Closure and Motor Impersistence of Eyelid in a Patient with Amyotrophic Lateral Sclerosis
- A Case of Frontotemporal Dementia with Amyotrophic Lateral Sclerosis Presenting with Pathological Gambling
- Psychosocial Responses and Quality of Life among Amyotrophic Lateral Sclerosis Patients and Their Caregivers
