Identification of Two Novel ANKRD11 Mutations: Highlighting Incomplete Penetrance in KBG Syndrome
- Affiliations
-
- 1Research Team in Genomics and Molecular Epidemiology of Genetic Diseases, Genomics Center of Human Pathologies, Faculty of Medicine and Pharmacy, University Mohammed V, Rabat, Morocco
- 2Department of Medical Genetics, National Institute of Health, Rabat, Morocco
- 3Medical Genetics Unit, CHU Ibn Sina, Rabat, Morocco
- 4Center of Consultations and External Explorations, HER, CHU Ibn Sina, Rabat, Morocco
- KMID: 2550232
- DOI: http://doi.org/10.3343/alm.2024.44.1.110
Figure
-
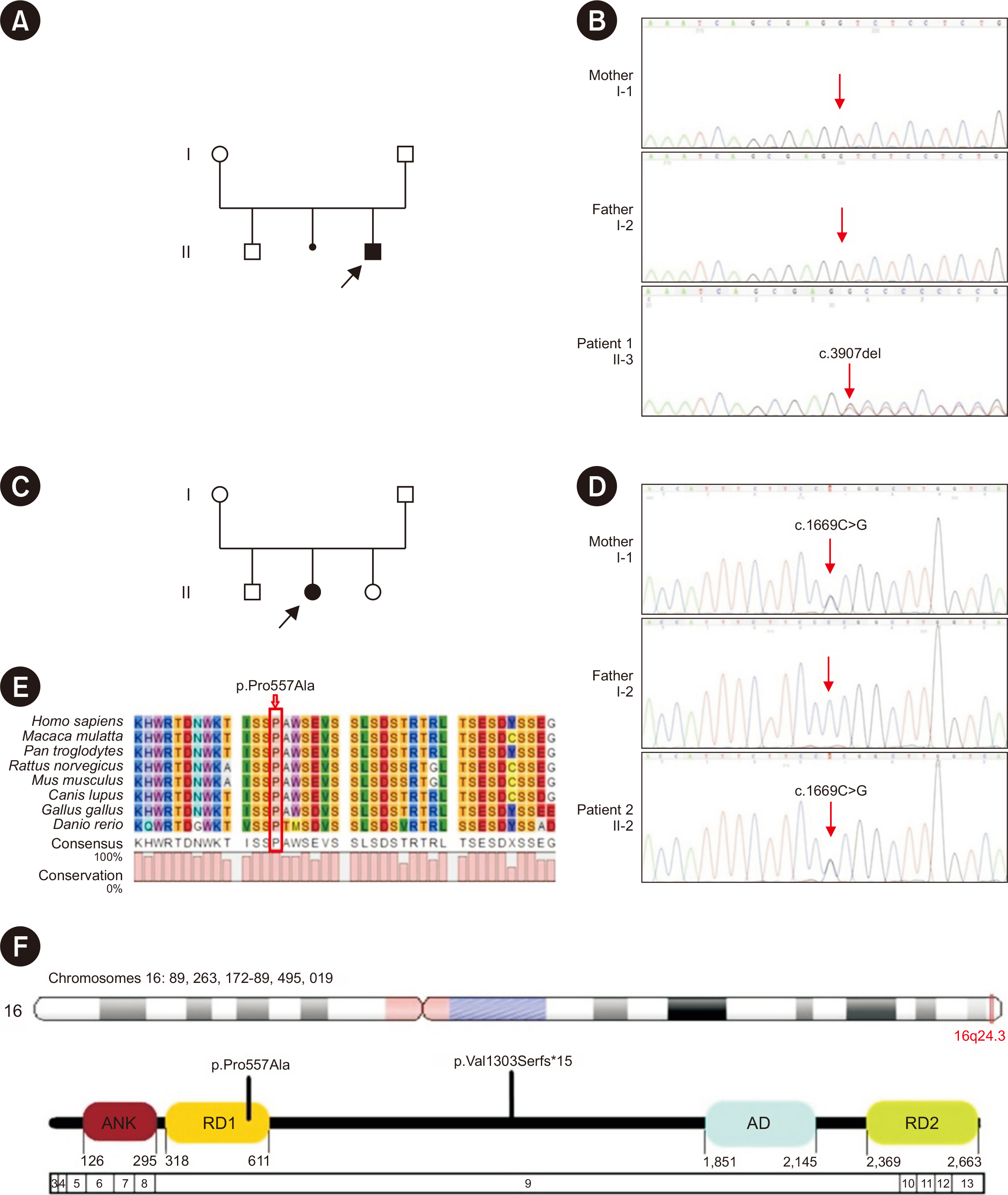
Fig. 1 Genetic analysis of the detected variants involved in our study. (A) The pedigree of patient 1’s family. (B) Electropherograms showing the monoallelic variant ANKRD11:c.3907del in patient 1 (II-3) and its absence in his parents (I-1 & I-2). (C) The pedigree of patient 2’s family. (D) Electropherograms showing the monoallelic mutation ANKRD11:c.1669C>G in patient 2 (II-2) and her mother (I-1) and its absence in her father (I-2). (E) Amino acid alignment of the ANKRD11 protein showing the high conservation of the residue Pro557 (highlighted by a red box) among different mammals. (F) The schematic diagram of the ANKRD11 protein structure showing the location of the two variants on protein level. ANKRD11 protein contains 2,663 residues and 4 functional domains: ANK: ankyrin domains, RD1: repression domain-1, AD: activation domain, RD2: repression domain-2. Patient 1 harbors p.Pro557Ala located on the RD1 domain. Patient 2 harbors p.Val1303Serfs*15 located between RD1 and AD domains.
Reference
-
1. Goldenberg A, Riccardi F, Tessier A, Pfundt R, Busa T, Cacciagli P, et al. 2016; Clinical and molecular findings in 39 patients with KBG syndrome caused by deletion or mutation of ANKRD11. Am J Med Genet A. 170:2847–59. DOI: 10.1002/ajmg.a.37878. PMID: 27605097.2. Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, et al. 2011; Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 89:289–94. DOI: 10.1016/j.ajhg.2011.06.007. PMID: 21782149. PMCID: PMC3155157.3. Zhang T, Yang Y, Yin X, Wang X, Ni J, Dong Z, et al. 2021; Two loss-of-function ANKRD11 variants in Chinese patients with short stature and a possible molecular pathway. Am J Med Genet A. 185:710–8. DOI: 10.1002/ajmg.a.62024. PMID: 33354850. PMCID: PMC7898801.4. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. 2015; Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17:405–24. DOI: 10.1038/gim.2015.30. PMID: 25741868. PMCID: PMC4544753.
Article5. Li Q, Sun C, Yang L, Lu W, Luo F. 2021; Comprehensive analysis of clinical spectrum and genotype associations in Chinese and literature reported KBG syndrome. Transl Pediatr. 10:834–42. DOI: 10.21037/tp-20-385. PMID: 34012832. PMCID: PMC8107870.
Article6. de Boer E, Ockeloen CW, Kampen RA, Hampstead JE, Dingemans AJM, Rots D, et al. 2022; Missense variants in ANKRD11 cause KBG syndrome by impairment of stability or transcriptional activity of the encoded protein. Genet Med. 24:2051–64. DOI: 10.1016/j.gim.2022.06.007. PMID: 35833929.7. Parenti I, Mallozzi MB, Hüning I, Gervasini C, Kuechler A, Agolini E, et al. 2021; ANKRD11 variants: KBG syndrome and beyond. Clin Genet. 100:187–200. DOI: 10.1111/cge.13977. PMID: 33955014.8. Scarano E, Tassone M, Graziano C, Gibertoni D, Tamburrino F, Perri A, et al. 2019; Novel mutations and unreported clinical features in KBG syndrome. Mol Syndromol. 10:130–8. DOI: 10.1159/000496172. PMID: 31191201. PMCID: PMC6528090.
Article9. Low K, Ashraf T, Canham N, Clayton-Smith J, Deshpande C, Donaldson A, et al. 2016; Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 170:2835–46. DOI: 10.1002/ajmg.a.37842. PMID: 27667800. PMCID: PMC5435101.
Article10. Murray N, Burgess B, Hay R, Colley A, Rajagopalan S, McGaughran J, et al. 2017; KBG syndrome: An Australian experience. Am J Med Genet A. 173:1866–77. DOI: 10.1002/ajmg.a.38121. PMID: 28449295.
Article11. Kim HJ, Cho E, Park JB, Im WY, Kim HJ. 2015; A Korean family with KBG syndrome identified by ANKRD11 mutation, and phenotypic comparison of ANKRD11 mutation and 16q24.3 microdeletion. Eur J Med Genet. 58:86–94. DOI: 10.1016/j.ejmg.2014.11.003. PMID: 25464108.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- An ANKRD11 exonic deletion accompanied by a congenital megacolon in an infant with KBG syndrome
- Early Diagnosis of KBG Syndrome Using Diagnostic Exome Sequencing
- A de novo Microdeletion of ANKRD11 Gene in a Korean Patient with KBG Syndrome
- Familial Spinal Muscular Atrophy wigh Autosomal Dominant Inheritance
- A Case of Late-Onset Li-Fraumeni-like Syndrome with Unilateral Breast Cancer
