The Outcome of Long QT Syndrome, a Korean Single Center Study
- Affiliations
-
- 1Department of Pediatrics, Seoul National University Children’s Hospital, Seoul National University College of Medicine, Seoul, Korea
- 2Department of Pediatrics, Gachon University Gil Medical Center, Incheon, Korea
- 3Department of Pediatrics, Sejong General Hospital, Bucheon, Korea
- 4Department of Internal Medicine, Seoul National University Children’s Hospital, Seoul National University College of Medicine, Seoul, Korea
- KMID: 2534041
- DOI: http://doi.org/10.4070/kcj.2022.0081
Abstract
- Background and Objectives
Although long QT syndrome (LQTS) is a potentially lifethreatening inherited cardiac channelopathy, studies documenting the long-term clinical data of Korean patients with LQTS are scarce.
Methods
This retrospective cohort study included 105 patients with LQTS (48 women; 45.7%) from a single tertiary center. The clinical outcomes were analyzed for the rate of freedom from breakthrough cardiac events (BCEs), additional treatment needed, and death.
Results
LQTS was diagnosed at a median age of 11 (range, 0.003–80) years. Genetic testing was performed on 90 patients (yield, 71.1%). The proportions of genetically confirmed patients with LQTS types 1, 2, 3, and others were 34.4%, 12.2%, 12.2%, and 12.2%, respectively. In the symptomatic group (n=70), aborted cardiac arrest was observed in 30% of the patients. Treatments included medications in 60 patients (85.7%), implantable cardioverter-defibrillators in 27 (38.6%; median age, 17 years; range, 2–79 years), and left cardiac sympathetic denervation surgery in 7 (10%; median age, 13 years; range, 2–34). The 10-year BCE-free survival rate was 73.2%. By genotype, significant differences were observed in BCEs despite medication (p<0.001). The 10-year BCE-free survival rate was the highest in patients with LQTS type 1 (81.8%) and the lowest in those with multiple LQTS-associated mutations (LQTM). All patients with LQTS survived, except for one patient who had LQTM.
Conclusions
Good long-term outcomes can be achieved by using recently developed genetically tailored management strategies for patients with LQTS.
Figure
-
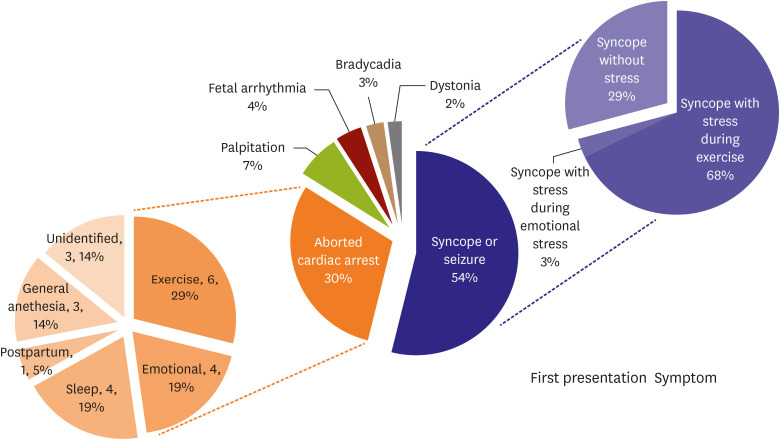
Figure 1 The mode of initial presentation in the symptomatic group.

Figure 2 The distributions of genetic mutations are confirmed in the genetically verified group.G−/P+ = genotype negative and phenotype positive; JLNS = Jervell Lange-Nielsen syndrome; LQT1 = congenital long QT syndrome type 1; LQT2 = congenital long QT syndrome type 2; LQT3 = congenital long QT syndrome type 3; LQT4 = congenital long QT syndrome type 4; LQT7 = congenital long QT syndrome type 7; LQT8 = congenital long QT syndrome type 8; LQTM = multiple long QT syndrome-associated mutations; VUS = variant of uncertain significance.
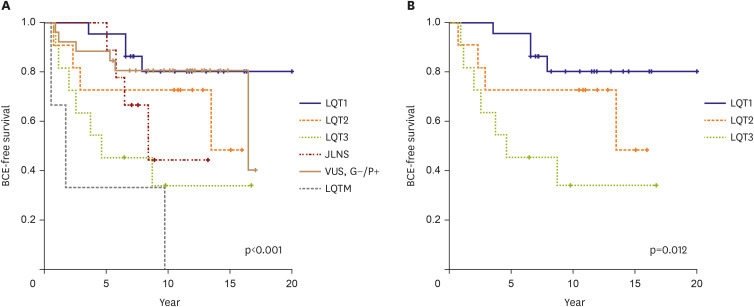
Figure 3 BCE-free survival curve for entire genetically verified LQTS. (A) BCE-free survival through each genotype, statistical significance was analyzed except for LQT4, 7, 8 which were rare. (B) When comparing LQT1, 2, and 3, which account for most of LQTS, the prognosis was poor in the order of LQT3, LQT2, and LQT1.BCE = breakthrough cardiac event; G−/P+ = genotype negative and phenotype positive; LQTS = long QT syndrome; LQT1 = congenital long QT syndrome type 1; LQT2 = congenital long QT syndrome type 2; LQT3 = congenital long QT syndrome type 3; LQT4 = congenital long QT syndrome type 4; LQT7 = congenital long QT syndrome type 7; LQT8 = congenital long QT syndrome type 8; LQTM = multiple LQTS-associated mutations; VUS = variant of uncertain significance.
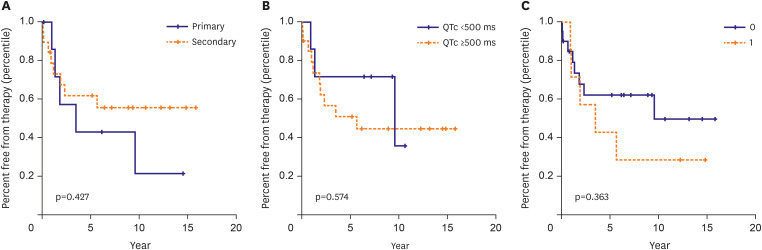
Figure 4 Individual/cumulative risk factors for appropriate ICD shock. Among the ICD recipients, the Kaplan-Meier curves were analyzed through the risk factors, primary vs. secondary prevention (A), QTc ≥500 ms (B), and family history of sudden cardiac death (C) for the event that received appropriate shock treatment.ICD = implantable cardioverter-defibrillator; QTc = corrected QT interval.
Cited by 2 articles
-
Multicenter Cohort Analysis Unveil Inherited Arrhythmia in Korea
Il-young Oh
Korean Circ J. 2023;53(10):708-709. doi: 10.4070/kcj.2023.0214.Clinical and Genetic Features of Korean Inherited Arrhythmia Probands
Joo Hee Jeong, Suk-Kyu Oh, Yun Gi Kim, Yun Young Choi, Hyoung Seok Lee, Jaemin Shim, Yae Min Park, Jun-Hyung Kim, Yong-Seog Oh, Nam-Ho Kim, Hui-Nam Pak, Young Keun On, Hyung Wook Park, Gyo-Seung Hwang, Dae-Kyeong Kim, Young-Ah Park, Hyoung-Seob Park, Yongkeun Cho, Seil Oh, Jong-Il Choi, Young-Hoon Kim
Korean Circ J. 2023;53(10):693-707. doi: 10.4070/kcj.2023.0083.
Reference
-
1. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015; 116:1887–1906. PMID: 26044246.2. Tester DJ, Ackerman MJ. Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J Am Coll Cardiol. 2007; 49:240–246. PMID: 17222736.3. Skinner JR, Crawford J, Smith W, et al. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm. 2011; 8:412–419. PMID: 21070882.4. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995; 80:795–803. PMID: 7889573.5. Napolitano C, Priori SG, Schwartz PJ, et al. Genetic testing in the long QT syndrome: development and validation of an efficient approach to genotyping in clinical practice. JAMA. 2005; 294:2975–2980. PMID: 16414944.6. Mizusawa Y, Horie M, Wilde AA. Genetic and clinical advances in congenital long QT syndrome. Circ J. 2014; 78:2827–2833. PMID: 25274057.7. Abriel H, Zaklyazminskaya EV. Cardiac channelopathies: genetic and molecular mechanisms. Gene. 2013; 517:1–11. PMID: 23266818.8. Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011; 124:2181–2184. PMID: 22083145.9. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013; 10:1932–1963. PMID: 24011539.10. Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012; 5:868–877. PMID: 22895603.11. Sundström E, Jensen SM, Diamant UB, Rydberg A. Implantable cardioverter defibrillator treatment in long QT syndrome patients: a national study on adherence to international guidelines. Scand Cardiovasc J. 2017; 51:88–94. PMID: 27936942.12. Bos JM, Bos KM, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation in long QT syndrome: analysis of therapeutic nonresponders. Circ Arrhythm Electrophysiol. 2013; 6:705–711. PMID: 23728945.13. Garson A Jr, Dick M 2nd, Fournier A, et al. The long QT syndrome in children. An international study of 287 patients. Circulation. 1993; 87:1866–1872. PMID: 8099317.14. Koponen M, Marjamaa A, Hiippala A, et al. Follow-up of 316 molecularly defined pediatric long-QT syndrome patients: clinical course, treatments, and side effects. Circ Arrhythm Electrophysiol. 2015; 8:815–823. PMID: 26063740.15. Rohatgi RK, Sugrue A, Bos JM, et al. Contemporary outcomes in patients with long QT syndrome. J Am Coll Cardiol. 2017; 70:453–462. PMID: 28728690.16. Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008; 117:2184–2191. PMID: 18427136.17. Lee YS, Kwon BS, Kim GB, et al. Long QT syndrome: a Korean single center study. J Korean Med Sci. 2013; 28:1454–1460. PMID: 24133349.18. Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006; 296:1249–1254. PMID: 16968849.19. Drici MD, Burklow TR, Haridasse V, Glazer RI, Woosley RL. Sex hormones prolong the QT interval and downregulate potassium channel expression in the rabbit heart. Circulation. 1996; 94:1471–1474. PMID: 8823008.20. Barsheshet A, Dotsenko O, Goldenberg I. Genotype-specific risk stratification and management of patients with long QT syndrome. Ann Noninvasive Electrocardiol. 2013; 18:499–509. PMID: 24206565.21. D’Adamo P, Fassone L, Gedeon A, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet. 1997; 61:862–867. PMID: 9382096.22. Seo SH, Kim SY, Cho SI, et al. Application of multigene panel sequencing in patients with prolonged rate-corrected QT interval and no pathogenic variants detected in KCNQ1, KCNH2, and SCN5A . Ann Lab Med. 2018; 38:54–58. PMID: 29071820.23. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000; 101:616–623. PMID: 10673253.24. Mazzanti A, Maragna R, Faragli A, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol. 2016; 67:1053–1058. PMID: 26940925.25. Niaz T, Bos JM, Sorensen KB, Moir C, Ackerman MJ. Left cardiac sympathetic denervation monotherapy in patients with congenital long QT syndrome. Circ Arrhythm Electrophysiol. 2020; 13:e008830. PMID: 33198487.26. Mazzanti A, Maragna R, Vacanti G, et al. Interplay between genetic substrate, QTc duration, and arrhythmia risk in patients with long QT syndrome. J Am Coll Cardiol. 2018; 71:1663–1671. PMID: 29650123.27. Horner JM, Kinoshita M, Webster TL, Haglund CM, Friedman PA, Ackerman MJ. Implantable cardioverter defibrillator therapy for congenital long QT syndrome: a single-center experience. Heart Rhythm. 2010; 7:1616–1622. PMID: 20816872.28. Schwartz PJ, Spazzolini C, Priori SG, et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them?: data from the European Long-QT Syndrome Implantable Cardioverter-Defibrillator (LQTS ICD) Registry. Circulation. 2010; 122:1272–1282. PMID: 20837891.29. Gaba P, Bos JM, Cannon BC, et al. Implantable cardioverter-defibrillator explantation for overdiagnosed or overtreated congenital long QT syndrome. Heart Rhythm. 2016; 13:879–885. PMID: 26681611.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Implantable Cardioverter-Defibrillator(ICD) Therapy in a Patient with the Long QT Syndrome
- Anesthetic Management of a Patient with Congenital Long QT Syndrome: A case report
- Cardiac Arrest under Anesthesia in a Child with Previously Undiagnosed Long QT Syndrome: A case report
- Fever-Induced QTc Prolongation and Ventricular Fibrillation in a Healthy Young Man
- A Case of Congenital Long QT Syndrome with Reccurent Syncope
