miR-4487 Enhances Gefitinib-Mediated Ubiquitination and Autophagic Degradation of EGFR in Non-Small Cell Lung Cancer Cells by Targeting USP37
- Affiliations
-
- 1Department of Oral Physiology, Institute of Biomaterial-Implant, School of Dentistry, Wonkwang University, Iksan, Korea
- 2Wonkwang Dental Research Institute, School of Dentistry, Wonkwang University, Iksan, Korea
- 3Department of Carbon Convergence Engineering, College of Engineering, Wonkwang University, Iksan, Korea
- 4Department of Internal Medicine, School of Medicine, Wonkwang University, Iksan, Korea
- KMID: 2528214
- DOI: http://doi.org/10.4143/crt.2021.622
Abstract
- Purpose
With the identification of epidermal growth factor receptor (EGFR) mutations in non–small cell lung cancer (NSCLC) cells, EGFR–tyrosine kinase inhibitors (TKIs) are being used widely as the first-line of treatment in NSCLC. These inhibitors block auto-phosphorylation of activated EGFR by competing with ATP binding and mediate EGFR degradation independent of exogenous epidermal growth factor, which is associated with the mutation variants of EGFR. However, the precise mechanisms underlying the TKI-mediated EGFR degradation are still unclear.
Materials and Methods
To examine the physiological roles of miR-4487 and ubiquitin-specific peptidase 37 (USP37) in gefitinib-mediated EGFR degradation in NSCLC cells, multiple NSCLC cell lines were applied. The level of EGFR expression, apoptosis marker and autophagic flux were determined by western blot. Expression level of miR-4487 and cell cycle arrest was analyzed by TaqMan assay and flow cytometry respectively.
Results
We found that gefitinib mediates EGFR degradation under normal culture conditions, and is dependent on autophagic flux and the mutation variants of EGFR. Gefitinib reduced expression levels of USP37, which mediated EGFR degradation similar to gefitinib. Our results also showed a gefitinib-mediated increase in endogenous miR-4487 level and presented evidence for the direct targeting of USP37 by miR-4487, resulting in the sequential enhancement of ubiquitination, autophagy, and EGFR degradation. Thus, the depletion of USP37 and overexpression of miR-4487 led to an increase in gefitinib-mediated apoptotic cell death.
Conclusion
These data suggest that miR-4487 is a potential target for treating NSCLC, and miR-4487/USP37-regulated EGFR degradation is a determinant for developing gefitinib resistance.
Figure
-
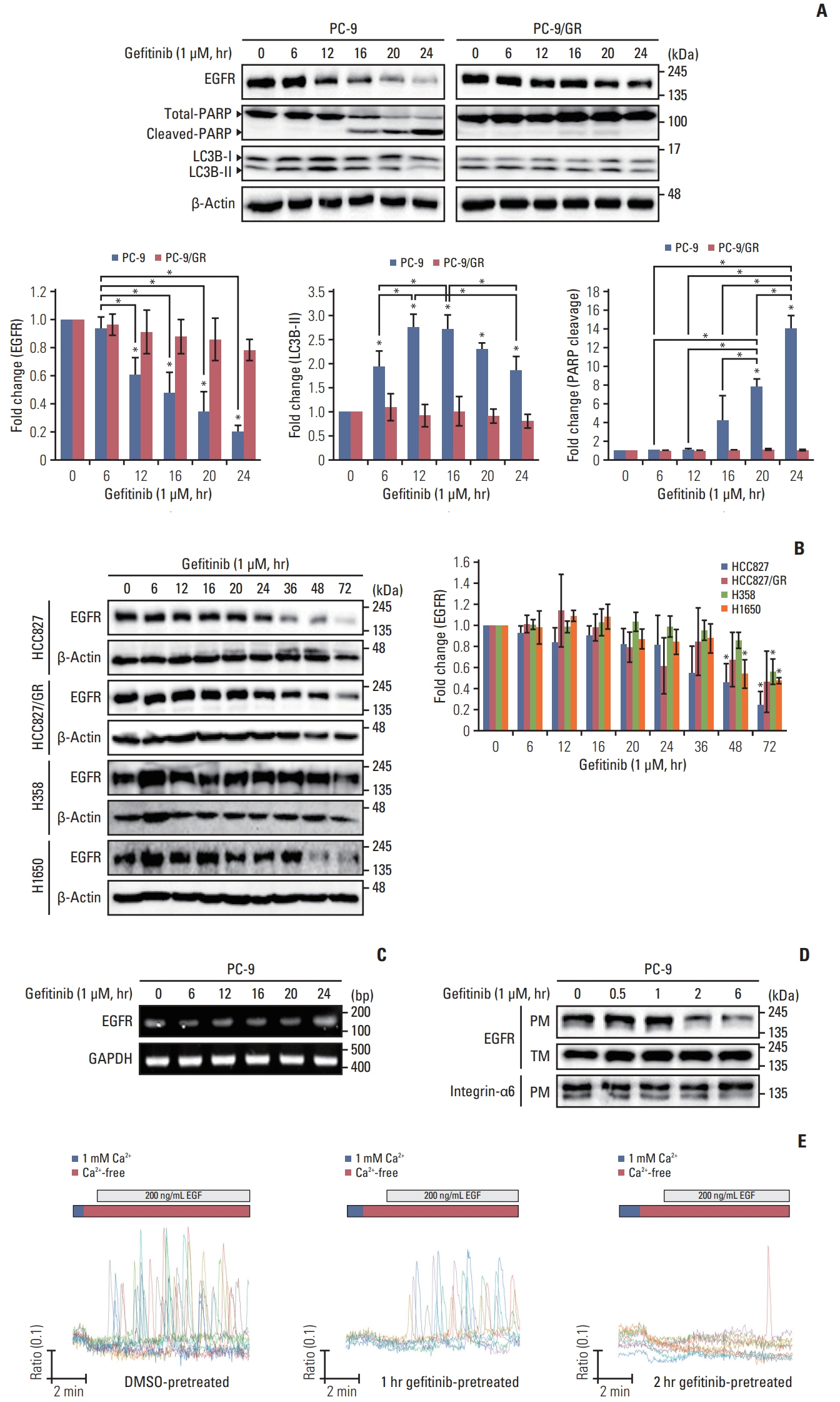
Fig. 1 Effects of long-term treatment of gefitinib on non–small cell lung cancer cells. Different cell lines were incubated with gefitinib (1 μM) for the indicated time. (A) Endogenous levels of epidermal growth factor receptor (EGFR) and cleavage of poly(ADP-ribose) polymerase-1 (PARP) and LC3B were determined using a western blot assay in the PC-9 and PC-9/GR cell lines. (B) Endogenous level of EGFR in HCC827, HCC827/GR, H358, and H1650 cells. β-Actin levels were used as loading controls. Column graphs show relative fold change of each protein in gefitinib-treated cells compared with that in control (0; no-gefitinib). Data are mean±standard deviation. *p < 0.05 compared with control or between indicated group. (C) Reverse transcription polymerase chain reaction was performed to detect the transcriptional activity of EGFR in gefitinib-treated PC-9 cells. (D) PC-9 cells were treated with gefitinib (1 μM) for the indicated time, and total membrane was fractionized and isolated proteins from each fraction were used to detect EGFR levels. Integrin-α6 was used as loading control. PM, plasma membrane; TM, total membrane. (E) Cells were pretreated with gefitinib (1 μM) before the experiments, and PC-9 cells loaded with Fura-2AM were used to determine intracellular Ca2+ ([Ca2+]i) response to epidermal growth factor (EGF; 200 ng/mL) in the absence of extracellular Ca2+. Each trace represents [Ca2+]i mobilization in a single cell.
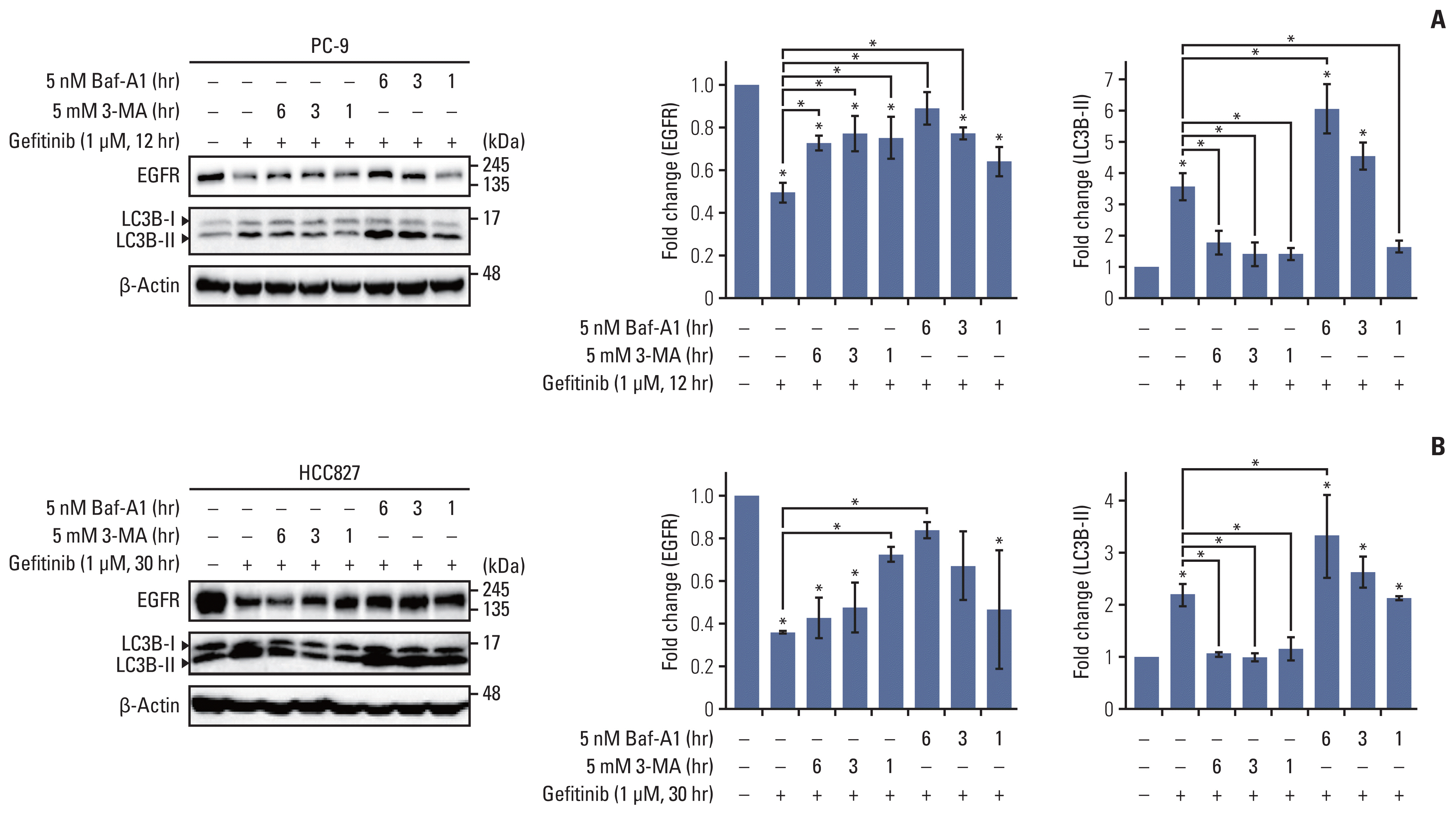
Fig. 2 Gefitinib-mediated epidermal growth factor receptor (EGFR) degradation is inhibited by autophagy inhibitors, 3-methyladenine (3-MA) and bafilomycin A1. Gefitinib (1μM)-exposed PC-9 (A) and HCC827 (B) cells were treated with bafilomycin A1 (5 nM) and 3-MA (5 mM) for 6 hours, 3 hours, and 1 hour before the end of incubation, and endogenous level of EGFR and LC3B cleavage were evaluated using western blot assay. Column graph data indicate fold change of each protein in inhibitor-treated cells compared with that in control (vehicle only). Data are represented as mean± standard deviation. * p < 0.05 compared with control or between indicated group.

Fig. 3 Endogenous level of ubiquitin-specific peptidase 37 (USP37) is down-regulated by gefitinib and USP37 depletion induces epidermal growth factor receptor (EGFR) degradation. (A) Endogenous levels of EGFR and LC3B cleavage were analyzed using western blot assay in BEAS-2B, PC-9, HCC827, H358, H1650, PC-9/GR, and HCC827/GR cell lines. Statistical data indicate fold change in USP37, LC3B cleavage in different cell lines compared with that in BEAS-2B cells. (B) Whole cell lysates isolated from gefitinib (1 μM)-treated PC-9 cells were used for western blot assay. Statistical data were presented as fold change of USP37 expression in cells (gefitinib-treated) compared to that in controls (no-gefitinib). Data are represented as mean± standard deviation. * p < 0.05 compared with control. (C) PC-9 cells were transfected with siUSP37 and then cultured with or without gefitinib (1 μM). After incubation, whole cell lysates were used for determining the endogenous levels of USP37, EGFR, LC3B cleavage, and ubiquitination.

Fig. 4 miR-4487 directly targets ubiquitin-specific peptidase 37 (USP37) and promotes epidermal growth factor receptor (EGFR) degradation and ubiquitination. (A) Differential expression of endogenous miR-4487 was evaluated in BEAS-2B and non–small cell lung cancer (NSCLC) cells using TaqMan primers. Statistical data were presented as fold change of miR-4487 levels in the tested cell lines compared to that in BEAS-2B (control) cells. Data are represented as mean±standard deviation (SD). *p < 0.05 compared with BEAS-2B cells. (B) Endogenous level of miR-4487 was measured in gefitinib (1 μM)-treated PC-9 cells using TaqMan primers. Statistical data were presented as fold change in miR-4487 levels in gefitinib-treated cells compared to that in control (0; no-gefitinib). Data are represented as mean±SD. *p < 0.05 compared with non–gefitinib-treated cells. (C) PC-9 and PC-9/GR cells were transfected with miR-4487 mimic and inhibitor. Total RNA was used for quantitative polymerase chain reaction analysis (left panel). The wild-type (WT) and mutant sequences of USP37 3′-untranslated region (3′-UTR) are listed for comparison. HEK293 cells were co-transfected with either WT or mutated 3′-UTR plasmid and with miR-4487 mimic. Luciferase activity was measured at 48 hours after transfection. Firefly luciferase activity was normalized to Renilla luciferase activity and presented as relative to control (Luc-WT USP37 3′-UTR w/o miR-4487). Data are represented as mean±SD. *p < 0.05 compared to control. I, miR-4487 inhibi tor; M, miR-4487 mimic; NC, negative control. (D) miR-4487 mimic and inhibitor was transfected into NSCLC cell lines and endogenous USP37 expression was determined using the western blot assay. (E) After transfection with NC, M, and I, PC-9 cells were exposed to gefitinib (1 μM) for 24 hours. The endogenous levels of EGFR and LC3B cleavage were analyzed using a western blot assay. (F) miR-4487 mimic was transfected into PC-9 cells and then exposed to gefitinib (1 μM) for 24 hours. Whole-cell ubiquitination was assessed using the UBI-QAPTURE-Q Kit.
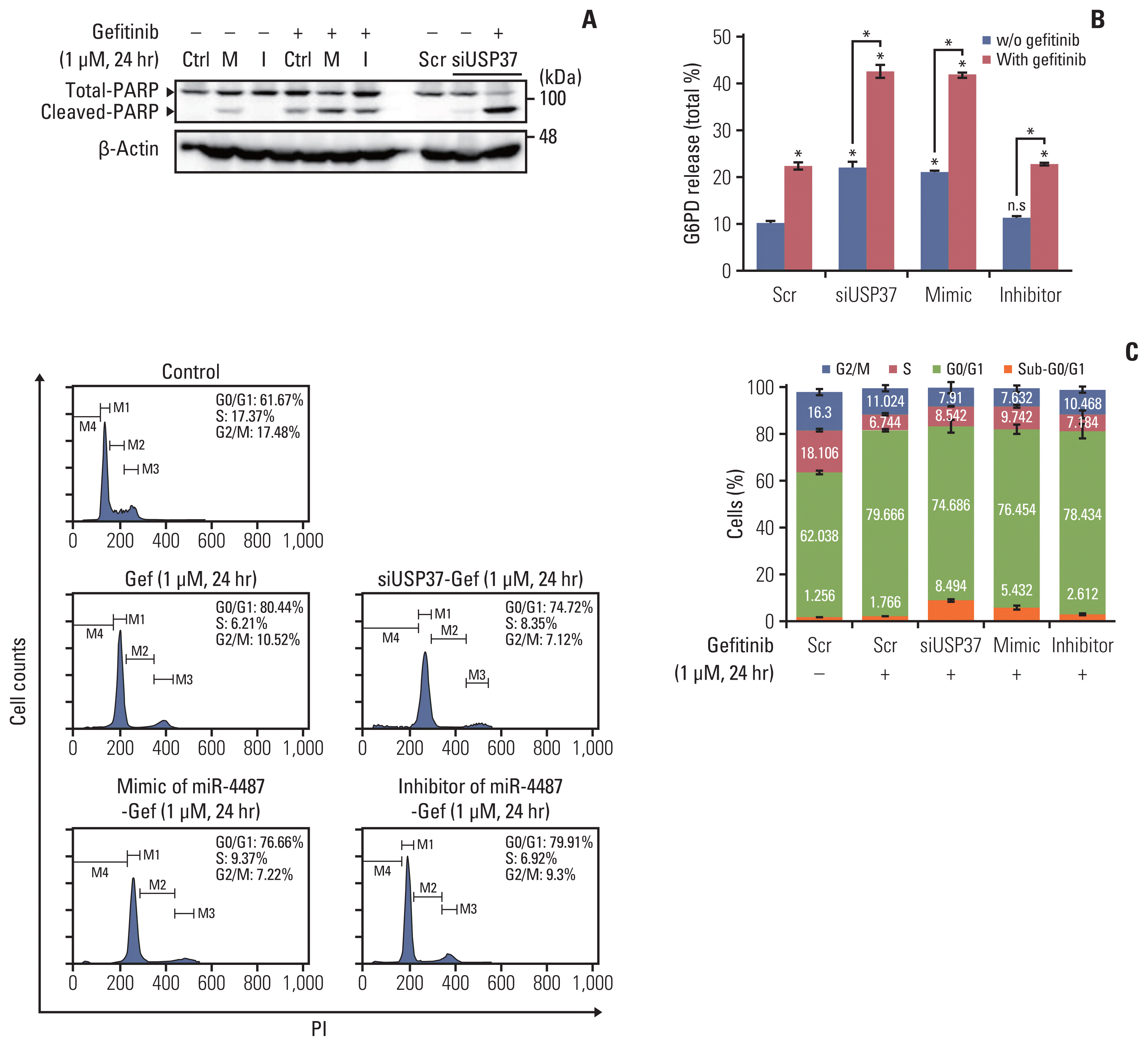
Fig. 5 Effects of miR-4487 overexpression and ubiquitin-specific peptidase 37 (USP37) depletion on gefitinib-mediated poly(ADP-ribose) polymerase-1 (PARP) cleavage, cytotoxicity, and apoptotic cell death. PC-9 cells were transfected with miR-4487 mimic (M)/inhibitor (I) and siUSP37 for 48 hours. Next, the cells were incubated for 24 hours with gefitinib (1 μM) and used to determine PARP cleavage (A), cytotoxicity (B), and cell cycle (C). (B) Cytotoxicity was evaluated by measuring glucose-6-phosphate dehydrogenase (G6PD) release. Data are presented as proportion of released G6PD (% total) and calculated as the mean±standard deviation. *p < 0.05 compared with scramble or between indicated groups. (C) Cells (2×104) stained with propidium iodide/RNase buffer were evaluated using flow cytometry. Data are represented as percentages and compared to those in control (scramble without gefitinib).
Reference
-
References
1. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018; 12:3–20.2. Zhou X, Zheng M, Chen F, Zhu Y, Yong W, Lin H, et al. Gefitinib inhibits the proliferation of pancreatic cancer cells via cell cycle arrest. Anat Rec (Hoboken). 2009; 292:1122–7.
Article3. Dragowska WH, Weppler SA, Wang JC, Wong LY, Kapanen AI, Rawji JS, et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS One. 2013; 8:e76503.
Article4. Liu Z, He K, Ma Q, Yu Q, Liu C, Ndege I, et al. Autophagy inhibitor facilitates gefitinib sensitivity in vitro and in vivo by activating mitochondrial apoptosis in triple negative breast cancer. PLoS One. 2017; 12:e0177694.
Article5. Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011; 6:e18691.
Article6. Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011; 13:495–504.
Article7. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ. 2013; 20:21–30.
Article8. Pinilla-Macua I, Grassart A, Duvvuri U, Watkins SC, Sorkin A. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. Elife. 2017; 6:e31993.
Article9. Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, et al. Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J. 2002; 21:2407–17.
Article10. Sacco JJ, Coulson JM, Clague MJ, Urbe S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life. 2010; 62:140–57.
Article11. Qin T, Li B, Feng X, Fan S, Liu L, Liu D, et al. Abnormally elevated USP37 expression in breast cancer stem cells regulates stemness, epithelial-mesenchymal transition and cisplatin sensitivity. J Exp Clin Cancer Res. 2018; 37:287.
Article12. Hong K, Hu L, Liu X, Simon JM, Ptacek TS, Zheng X, et al. USP37 promotes deubiquitination of HIF2alpha in kidney cancer. Proc Natl Acad Sci U S A. 2020; 117:13023–32.13. Pan J, Deng Q, Jiang C, Wang X, Niu T, Li H, et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene. 2015; 34:3957–67.
Article14. Cai J, Li M, Wang X, Li L, Li Q, Hou Z, et al. USP37 promotes lung cancer cell migration by stabilizing snail protein via deubiquitination. Front Genet. 2019; 10:1324.
Article15. Pascut D, Krmac H, Gilardi F, Patti R, Calligaris R, Croce LS, et al. A comparative characterization of the circulating miR-Nome in whole blood and serum of HCC patients. Sci Rep. 2019; 9:8265.
Article16. Chen Y, Wang S, Zhang L, Xie T, Song S, Huang J, et al. Identification of ULK1 as a novel biomarker involved in miR-4487 and miR-595 regulation in neuroblastoma SH-SY5Y cell autophagy. Sci Rep. 2015; 5:11035.
Article17. Ono M, Hirata A, Kometani T, Miyagawa M, Ueda S, Kinoshita H, et al. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004; 3:465–72.
Article18. Ray P, Raghunathan K, Ahsan A, Allam US, Shukla S, Basrur V, et al. Ubiquitin ligase SMURF2 enhances epidermal growth factor receptor stability and tyrosine-kinase inhibitor resistance. J Biol Chem. 2020; 295:12661–73.
Article19. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010; 141:1117–34.
Article20. Chung BM, Raja SM, Clubb RJ, Tu C, George M, Band V, et al. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol. 2009; 10:84.21. Henriksen L, Grandal MV, Knudsen SL, van Deurs B, Grovdal LM. Internalization mechanisms of the epidermal growth factor receptor after activation with different ligands. PLoS One. 2013; 8:e58148.
Article22. Ray P, Tan YS, Somnay V, Mehta R, Sitto M, Ahsan A, et al. Differential protein stability of EGFR mutants determines responsiveness to tyrosine kinase inhibitors. Oncotarget. 2016; 7:68597–613.
Article23. Stark MS, Klein K, Weide B, Haydu LE, Pflugfelder A, Tang YH, et al. The prognostic and predictive value of melanoma-related microRNAs using tissue and serum: a microRNA expression analysis. EBioMedicine. 2015; 2:671–80.
Article24. Hu L, Zhang R, Yuan Q, Gao Y, Yang MQ, Zhang C, et al. The emerging role of microRNA-4487/6845-3p in Alzheimer’s disease pathologies is induced by Abeta25-35 triggered in SH-SY5Y cell. BMC Syst Biol. 2018; 12:119.25. Chen S, Li X, Feng J, Chang Y, Wang Z, Wen A. Autophagy facilitates the lapatinib resistance of HER2 positive breast cancer cells. Med Hypotheses. 2011; 77:206–8.
Article26. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005; 171:603–14.
Article27. Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009; 33:505–16.
Article28. Dobson TH, Hatcher RJ, Swaminathan J, Das CM, Shaik S, Tao RH, et al. Regulation of USP37 expression by REST-associated G9a-dependent histone methylation. Mol Cancer Res. 2017; 15:1073–84.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Inhibition of Ubiquitin-specific Peptidase 8 Suppresses Growth of Gefitinib-resistant Non-small Cell Lung Cancer Cells by Inducing Apoptosis
- Afatinib Mediates Autophagic Degradation of ORAI1, STIM1, and SERCA2, Which Inhibits Proliferation of Non–Small Cell Lung Cancer Cells
- Overcoming the Intrinsic Gefitinib-resistance via Downregulation of AXL in Non-small Cell Lung Cancer
- Picropodophyllotoxin Inhibits Cell Growth and Induces Apoptosis in Gefitinib-Resistant Non-Small Lung Cancer Cells by Dual-Targeting EGFR and MET
- Enhanced Sensitivity to Gefitinib after Radiation in Non-Small Cell Lung Cancer Cells
