A Novel VPS33B Variant Identified by Exome Sequencing in a Patient with Arthrogryposis-Renal Dysfunction-Cholestasis Syndrome
- Affiliations
-
- 1Department of Pediatrics, Korea University College of Medicine, Seoul, Korea. shimjo@korea.ac.kr
- KMID: 2462099
- DOI: http://doi.org/10.5223/pghn.2019.22.6.581
Abstract
- Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome is a rare autosomal recessive multisystemic disease that is associated with the liver, kidney, skin, and central nervous and musculoskeletal systems. ARC occurs as a result of mutations in the VPS33B (Vacuolar protein sorting 33 homolog B) or VIPAR (VPS33B interacting protein, apical-basolateral polarity regulator) genes. A female infant presented with neonatal cholestasis with a severe clinical outcome. She was diagnosed with ARC syndrome using targeted exome sequencing (TES). Exome sequencing revealed compound heterozygous mutations, c.707A>T and c.239+5G>A, in VPS33B, where c.707A>T was a novel variant; the resultant functional protein defects were predicted via in silico analysis. c.239+5G>A, a pathogenic mutation that affects splicing, is found in less than 0.1% of the general population. Invasive techniques, such as liver biopsies, did not contribute to a differential diagnosis of ARC syndrome; thus, early TES together with clinical presentations constituted an apparently accurate diagnostic procedure.
Keyword
MeSH Terms
Figure
-
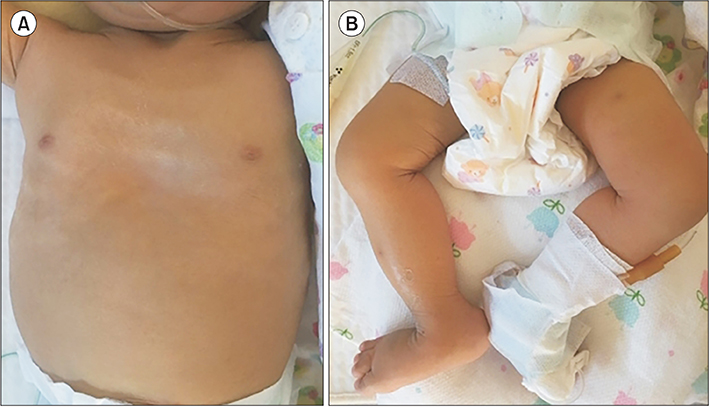
Fig. 1 The dysmorphic features associated with arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. An infant with ARC syndrome presenting symptoms of (A) ichthyosis and (B) arthrogryposis manifested by bilaterally dislocated hips, flexion contracture of the knees, and vertical talus.

Fig. 2 Micrographs from the liver biopsy of arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome patient. Micrographs from the histological examination (liver biopsy) of patient with ARC syndrome showing (A) giant cell hepatitis, (B) intrahepatic hematopoiesis, (C) lipofuscin deposition, and (D, E) a paucity of bile ducts (A-D: Sections were stained with hematoxylin and eosin and viewed under 400×magnification; E: Sections were stained with CK19 and viewed under 200×magnification).
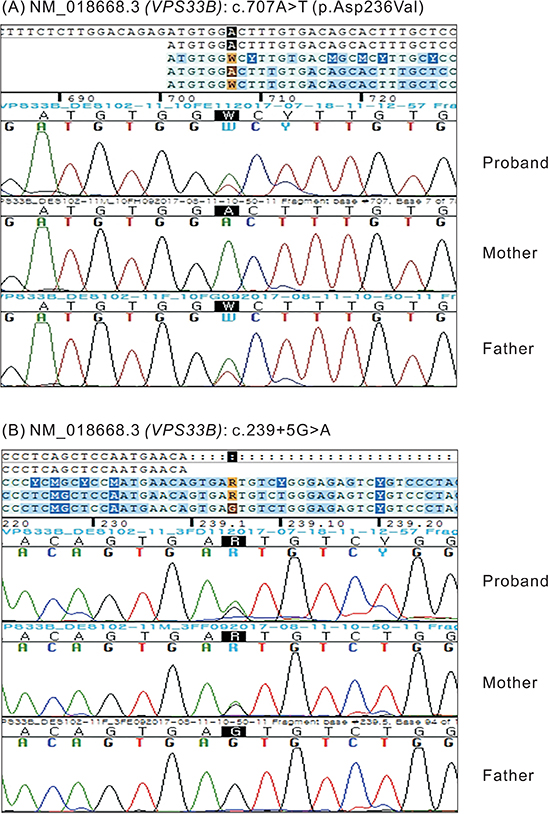
Fig. 3 Missense mutations found in a neonate with cholestasis. Molecular analysis of a neonate with cholestasis revealed compound heterozygous mutations in vacuolar protein sorting 33 homolog B. The DNA chromatograms highlighting the missense mutations: (A) c.707A>T (p.Asp236Val) and (B) c.239+5G>A. The c.707A>T mutation was a novel variant that was acquired from the father.
Reference
-
1. Lutz-Richner AR, Landolt RF. Familiare gallengansmissbildungen mit tubularer neireninsurfizienz. Helv Paediatr Acta. 1973; 28:1–12.2. Zhou Y, Zhang J. Arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome: from molecular genetics to clinical features. Ital J Pediatr. 2014; 40:77.
Article3. Gissen P, Tee L, Johnson CA, Genin E, Caliebe A, Chitayat D, et al. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006; 120:396–409.
Article4. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 531210 [Preprint]. 2019. cited 2019 Jan 28. Available from: http://dx.doi.org/10.1101/531210.5. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291.
Article6. 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO. A global reference for human genetic variation. Nature. 2015; 526:68–74.7. Saraiva JM, Lemos C, Gonçalves I, Carneiro F, Mota HC. Arthrogryposis multiplex congenita with renal and hepatic abnormalities in a female infant. J Pediatr. 1990; 117:761–763.
Article8. Di Rocco M, Callea F, Pollice B, Faraci M, Campiani F, Borrone C. Arthrogryposis, renal dysfunction and cholestasis syndrome: report of five patients from three Italian families. Eur J Pediatr. 1995; 154:835–839.
Article9. Coleman RA, Van Hove JL, Morris CR, Rhoads JM, Summar ML. Cerebral defects and nephrogenic diabetes insipidus with the ARC syndrome: additional findings or a new syndrome (ARCC-NDI)? Am J Med Genet. 1997; 72:335–338.
Article10. Nezelof C, Dupart MC, Jaubert F, Eliachar E. A lethal familial syndrome associating arthrogryposis multiplex congenita, renal dysfunction, and a cholestatic and pigmentary liver disease. J Pediatr. 1979; 94:258–260.
Article11. Horslen SP, Quarrell OW, Tanner MS. Liver histology in the arthrogryposis multiplex congenita, renal dysfunction, and cholestasis (ARC) syndrome: report of three new cases and review. J Med Genet. 1994; 31:62–64.
Article12. Mikati MA, Barakat AY, Sulh HB, Der Kaloustian VM. Renal tubular insufficiency, cholestatic jaundice, and multiple congenital anomalies--a new multisystem syndrome. Helv Paediatr Acta. 1984; 39:463–471.13. Eastham KM, McKiernan PJ, Milford DV, Ramani P, Wyllie J, Van't Hoff W, et al. ARC syndrome: an expanding range of phenotypes. Arch Dis Child. 2001; 85:415–420.
Article14. Ilhan O, Ozer EA, Ozdemir SA, Akbay S, Memur S, Kanar B, et al. Arthrogryposis-renal tubular dysfunction-cholestasis syndrome: a cause of neonatal cholestasis. case report. Arch Argent Pediatr. 2016; 114:e9–12.
Article15. Knisely AS. Progressive familial intrahepatic cholestasis in children. In : Dhawan A, editor. Concise pediatric and adolescent hepatology. pediatric and adolescent medicine 16. Basel: Karger;2012. p. 30–37.16. Carim L, Sumoy L, Andreu N, Estivill X, Escarceller M. Cloning, mapping and expression analysis of VPS33B, the human orthologue of rat Vps33b. Cytogenet Cell Genet. 2000; 89:92–95.
Article17. Cullinane AR, Straatman-Iwanowska A, Zaucker A, Wakabayashi Y, Bruce CK, Luo G, et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet. 2010; 42:303–312.
Article18. Gissen P, Johnson CA, Morgan NV, Stapelbroek JM, Forshew T, Cooper WN, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004; 36:400–404.
Article19. Matthews RP, Plumb-Rudewiez N, Lorent K, Gissen P, Johnson CA, Lemaigre F, et al. Zebrafish vps33b, an ortholog of the gene responsible for human arthrogryposis-renal dysfunction-cholestasis syndrome, regulates biliary development downstream of the onecut transcription factor hnf6. Development. 2005; 132:5295–5306.
Article20. Smith H, Galmes R, Gogolina E, Straatman-Iwanowska A, Reay K, Banushi B, et al. Associations among genotype, clinical phenotype, and intracellular localization of trafficking proteins in ARC syndrome. Hum Mutat. 2012; 33:1656–1664.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Two Cases of ARC (Arthrogryposis, renal dysfunction and cholestasis) Syndrome
- A Case of ARC (Arthrogryposis, Renal dysfunction and Cholestasis) Syndrome with a Dead Sibling Presenting Cholestatic Jaundice
- Clinical Characteristics of Arthrogryposis, Renal Tubular Dysfunction, Cholestasis(ARC) Syndrome in Korea
- A Case of Neonatal Cholestasis with Arthrogryposis Multiplex Congenita and Renal Tubular Insufficiency(ARC Syndrome)
- Exome Sequencing in Mendelian Disorders
