Brief introduction to current pharmacogenomics research tools
- Affiliations
-
- 1Department of Pharmacology and PharmacoGenomics Research Center, Inje University College of Medicine, Busan 47392, Korea. hosuegi@gmail.com
- 2Department of Clinical Pharmacology, Inje University Busan Paik Hospital, Busan 47392, Korea.
- KMID: 2383595
- DOI: http://doi.org/10.12793/tcp.2016.24.1.13
Abstract
- There is increasing interest in the application of personalized therapy to healthcare to increase the effectiveness of and reduce the adverse reactions to treatment. Pharmacogenomics is a core element in personalized therapy and pharmacogenomic research is a growing field. Understanding pharmacogenomic research tools enables better design, conduct, and analysis of pharmacogenomic studies, as well as interpretation of pharmacogenomic results. This review provides a general and brief introduction to pharmacogenomics research tools, including genotyping technology, web-based genome browsers, and software for haplotype analysis.
Keyword
Figure
-
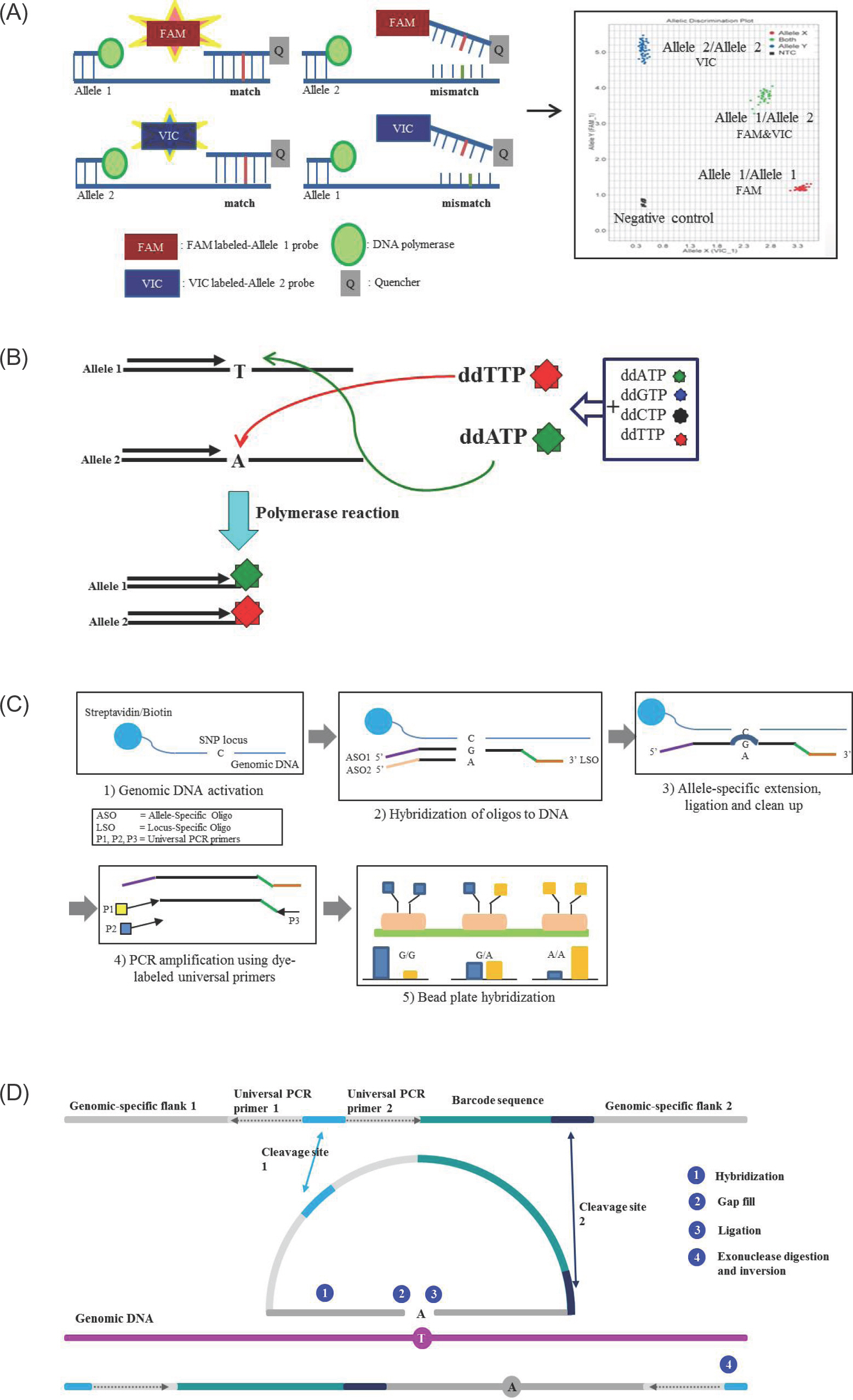
Figure 1. Schematic representation of SNP detection method. (A) TaqMan probe method. The template DNA is combined with primers and fluorophore-labeled allele specific probes, such as FAM labeled-allele 1 probe and VIC labeled-allele 2 probe. When a FAM-labeled allele 1 probe perfectly complements the target SNP site at allele 1, the FAM is released by the 5' nuclease activity of Taq polymerase. Release of the FAM separates the 3' quencher, allowing FAM to be emitted and subsequently detected as homozygotes of SNP at allele 1. In contrast, the VIC signal indicates homozygotes of SNP at allele 2. Fluorescences from both signals indicate heterozygotes. (B) Single base extension method. Extension primers are designed a single base upstream from the target SNP. During polymerase reactions, extension primers are bound a single base upstream of SNP site and ddATP are bound and extended to the target SNP site at allele 1, and the reaction is terminated. In contrast, ddTTP are bound and terminated to the target SNP site at allele 2. The incorporated base is detected using fluorescence. (C) Goldengate assay. Genomic DNA is activated by binding streptavidin/biotin beads. Both primers (ASOs and LSO) are hybridized to the genomic DNA-bound streptavidin/biotin beads. Extension of the appropriate ASO and ligation of the LSO generates ligation products. This product is amplified using dye-labeled universal PCR primers, and then fluorescence is used for signal detection. (D) GeneChip Microarray. ① Two genomic-specific flank regions are hybridized at genomic DNA ② The gap is filled with complementary base of target SNP and ③ ligated ④ cleavage site is digested by exonuclease, then the inversion probe is amplified by PCR reaction.

Figure 2. An example of what a Linkage Disequilibrium (LD) Map looks like (triangle format). This is a Linkage disequilibrium (LD) blocks structure of the inflammatory bowel disease 5 (IBD5) gene in chromosome 5q31-q33. The white line on top represents a strand of a chromosome. The black bars on the white line of the chromosome are SNPs (Single nucleotide polymorphism) that have been identified and sequenced. These SNP locations or loci are labeled in this picture as 1, 2, 3 and so on (#1∼20 in this case). The kilobase (kb) in each LD blocks means the distance between first of SNP and end of SNP. The values in diamond represent the D' values (×100) between the two SNPs. For example, the diamond in which the columns leading from SNP#1 and SNP#7 intersect has a number, 95 with red color. Thus SNP#1 and SNP#7 have a D' value of 0.95 and are in high linkage disequilibrium with each other. The color is categorized according to D' value (D' ≥ 0.80, red; 0.5 ≤ D' < 0.8, pink; 0.2 ≤ D' < 0.5, blue; and D' < 0.2, white).
Reference
-
References
1. Wang L, McLeod HL, Weinshilboum RM. Genomics and drug response. N Engl J Med. 2011; 364:1144–1153.
Article2. Mooney SD. Progress towards the integration of pharmacogenomics in practice. Hum Genet. 2015; 134:459–465.
Article3. Flynn AA. Pharmacogenetics: practices and opportunities for study design and data analysis. Drug Discov Today. 2011; 16:862–866.
Article4. Roden DM, George AL Jr. The genetic basis of variability in drug responses. Nat Rev Drug Discov. 2002; 1:37–44.
Article5. Lee JK, Part3 Disease association study. Genetic variation and Diseases (Language in Korean). 2nd ed.Seoul;2010. p. 211–262.6. Kim S, Misra A. SNP genotyping: technologies and biomedical applications. Annu Rev Biomed Eng. 2007; 9:289–320.
Article7. NwanKwo DC AM. Restriction enzymes and their uses in specific sequencing to produce predictable fragment of DNA making genetic engineering simply. Journal of Pharmaceutical Research and Opinion. 2011; 5:148–152.8. Nobile C, Romeo G. Partial digestion with restriction enzymes of ultraviolet-irradiated human genomic DNA: a method for identifying restriction site polymorphisms. Genomics. 1988; 3:272–274.
Article9. Koch WH. Technology platforms for pharmacogenomic diagnostic assays. Nat Rev Drug Discov. 2004; 3:749–761.
Article10. Livak KJ. SNP genotyping by the 5'-nuclease reaction. Methods Mol Biol. 2003; 212:129–147.
Article11. Frederickson RM. Fluidigm. Biochips get indoor plumbing. Chem Biol. 2002; 9:1161–1162.12. Hsiao SJ, Rai AJ, Multiplexed Pharmacogenetic Assays for SNP Genotyping: Tools and Techniques for Individualizing Patient Therapy. In: Dr. Des-pina Sanoudou (ed) Clinical Applications. 1st ed.Yan An Road (West), Shanghai;2012. p. 35–54.13. Nikolausz M, Chatzinotas A, Táncsics A, Imfeld G, Kästner M. The single-nucleotide primer extension (SNuPE) method for the multiplex detection of various DNA sequences: from detection of point mutations to microbial ecology. Biochem Soc Trans. 2009; 37:454–459. doi: 10.1042/BST0370454.
Article14. Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001; 11:3–11.
Article15. Zhou Z, Poe AC, Limor J, Grady KK, Goldman I, McCollum AM, et al. Pyrosequencing, a high-throughput method for detecting single nucleotide polymorphisms in the dihydrofolate reductase and dihydropteroate synthetase genes of Plasmodium falciparum. J Clin Microbiol. 2006; 44:3900–3910.16. Hurst CD, Zuiverloon TC, Hafner C, Zwarthoff EC, Knowles MA. A SNaPshot assay for the rapid and simple detection of four common hotspot codon mutations in the PIK3CA gene. BMC Res Notes. 2009; 2:66. doi: 10.1186/1756-0500-2-66.
Article17. Syrmis MW, Moser RJ, Kidd TJ, Hunt P, Ramsay KA, Bell SC, et al. High-throughput single389 nucleotide polymorphism-based typing of shared Pseudomonas aeruginosa strains in cystic fibrosis patients using the Sequenom iPLEX platform. J Med Microbiol. 2013; 62:734–740.18. Gabriel S, Ziaugra L, Tabbaa D. SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet. 2009. ;Chapter 2: Unit 2.12.doi: 10.1002/0471142905.hg0212s60.
Article19. Tian HL, Wang FG, Zhao JR, Yi HM, Wang L, Wang R, et al. Development of maizeSNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties. Mol Breed. 2015; 35:136.
Article20. Dalma-Weiszhausz DD, Warrington J, Tanimoto EY, Miyada CG. The affymetrix GeneChip platform: an overview. 398 Methods Enzymol. 2006; 410:3–28.21. Spudich GM, Fernández-Suárez XM. Touring Ensembl: a practical guide to genome browsing. BMC Genomics. 2010; 11:295. doi: 10.1186/1471-2164-11-295.
Article22. Wang J, Kong L, Gao G, Luo J. A brief introduction to web-based genome browsers. Brief Bioinform. 2013; 14:131–143.
Article23. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. doi: 10.1093/nar/gkt1113.
Article24. Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015; 43:D670–D681.25. Harel A, Inger A, Stelzer G, Strichman-Almashanu L, Dalah I, Safran M, et al. GIFtS: annotation landscape analysis with GeneCards. BMC Bioinformatics. 2009; 10:348.
Article26. Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, et al. GeneCards Version 3: the human gene integrator. Database (Oxford). 2010. baq020. doi: 010.1093/database/baq1020.
Article27. Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2014. Nucleic Acids Res. 2014; 42:D749–D755.
Article28. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012; 491:56–65.
Article29. Consortium IH. The International HapMap Project. Nature. 2003; 426:789–796.
Article30. Consortium IH. A haplotype map of the human genome. Nature. 2005; 437:1299–1320.
Article31. Clark AG. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol. 1990; 7:111–122.32. Barrett JC. Haploview: Visualization and analysis of SNP genotype data. Cold Spring Harb Protoc. 2009; 2009:pdb ip71.doi: 10.1101/pdb.ip1171.
Article33. Hawley ME, Kidd KK. HAPLO: a program using the EM algorithm to estimate the frequencies of multisite haplotypes. J Hered. 1995; 86:409–411.
Article34. Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995; 12:921–927.35. Clayton D. A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet. 1999; 65:1170–1177.
Article36. Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001; 68:978–989.
Article37. Morris AP, Whittaker JC, Balding DJ. Fine-scale mapping of disease loci via shattered coalescent modeling of genealogies. Am J Hum Genet. 2002; 70:686–707.
Article38. Niu T, Qin ZS, Xu X, Liu JS. Bayesian haplotype inference for multiple linked single434 nucleotide polymorphisms. Am J Hum Genet. 2002; 70:157–169.39. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005; 21:263–265.
Article40. Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003; 73:1162–1169.
Article41. Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005; 76:449–462.
Article42. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81:559–575.
Article
