Korean Circ J.
2017 Mar;47(2):278-281. 10.4070/kcj.2016.0321.
Identification of a Novel GLA Mutation (L206 P) in a Patient with Fabry Disease
- Affiliations
-
- 1Division of Cardiology, Department of Internal Medicine, St. Vincent's Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea. jiheekim@catholic.ac.kr
- 2Division of Nephrology, Department of Internal Medicine, St. Vincent's Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- 3Department of Ophthalmology, St. Vincent's Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- 4Department of Dermatology, St. Vincent's Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- 5Department of Pathology, St. Vincent's Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- KMID: 2377470
- DOI: http://doi.org/10.4070/kcj.2016.0321
Abstract
- We report a new α-Galactosidase A (αGal-A) mutation in a 39-year-old Korean born, male Fabry disease patient. Fabry disease is a devastating, progressive inborn error of metabolism caused by X-linked genetic mutations. In this case, the first clinical symptom to occur was in childhood consisting of a burning pain originating in the extremities then radiating inwards to the limbs. This patient also stated to have ringing in his ears, angiokeratomas on his trunk, and cornea verticillata. He visited an outpatient cardiologist due to intermittent and atypical chest discomfort at the age of 39. Electrocardiographic and echocardiographic examination showed left ventricular hypertrophy. A physical examination revealed proteinuria without hematuria. The patient's plasma αGal-A activity was markedly lower than the mean value of the controls. After genetic counseling and obtaining written informed consent, we identified one hemizygous mutation in exon 4 of galactosidase alpha, c.617T>C (p.Leu206 Pro). He was eventually diagnosed as having Fabry disease.
MeSH Terms
Figure
-

Fig. 1 At the initial outpatient visit, the patient's electrocardiogram (A) demonstrated marked left ventricular hypertrophy and an inverted T wave. The cardiac echocardiography (B) showed left ventricular hypertrophy.
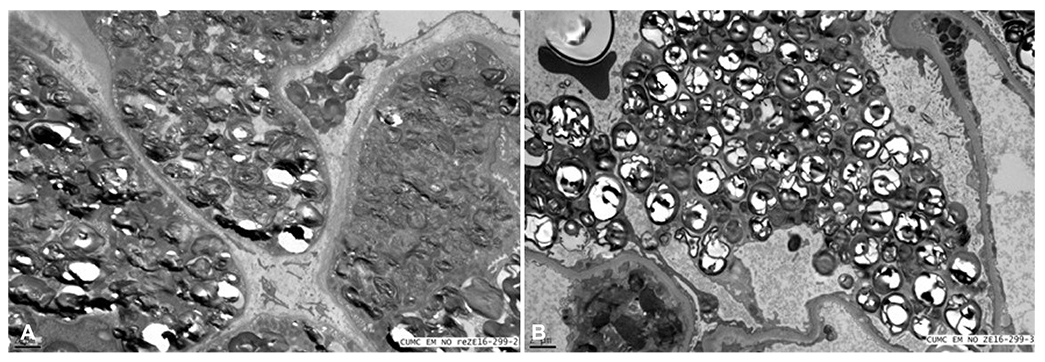
Fig. 2 The patient showed the renal pathology. (A) Glomeruli examined by electron microscopy show characteristic round, whorled, laminated electrondense structures filling the podocyte cytoplasm and a few endothelial cells. (B) The characteristic lipid inclusions with whorled architecture pack the tubular epithelial cytoplasm upon electron microscopy.
Reference
-
1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010; 5:30.2. Branton MH, Schiffmann R, Sabnis SG, et al. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002; 81:122–138.3. Umeda T, Hashimoto S, Noriyasu K, Takamura A, Fujisaki M, Eto Y. Identification of a novel GLA mutation (F69 L) in a Japanese patient with late-onset Fabry disease. Hum Genome Var. 2015; 2:15044.4. Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004; 34:236–242.5. Duve C. Exploring cells with a centrifuge. Science. 1975; 189:186–194.6. Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008; 105:2812–2817.7. Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008; 64:550–555.8. Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008; 93:112–128.9. Schiffmann R, Warnock DG, Banikazemi M, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009; 24:2102–2111.10. Naleschinski D, Arning K, Baron R. Fabry disease--pain doctors have to find the missing ones. Pain. 2009; 145:10–11.11. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999; 281:249–254.12. Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006; 79:31–40.13. Inoue T, Hattori K, Ihara K, Ishii A, Nakamura K, Hirose S. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet. 2013; 58:548–552.14. Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006; 8:539–548.15. Lee K, Jin X, Zhang K, et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology. 2003; 13:305–313.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Two cases of Fabry disease identified in brothers
- A Novel Frameshift Mutation of Galactosidase-alpha in Fabry Disease Restricted to Dermatologic Manifestations
- Fabry disease screening in young patients with acute ischemic stroke in Korea
- Effects of a chemical chaperone on genetic mutations in alpha-galactosidase A in Korean patients with Fabry disease
- A novel GLA mutation in a Korean boy with an early cardiac manifestation of Fabry disease
