Recently Identified Forms of Epidermolysis Bullosa
- Affiliations
-
- 1St John's Institute of Dermatology, King's College London (Guy's Campus), London, UK. john.mcgrath@kcl.ac.uk
- KMID: 2157440
- DOI: http://doi.org/10.5021/ad.2015.27.6.658
Abstract
- Epidermolysis bullosa (EB) comprises a collection of clinically diverse inherited blistering diseases that affect the skin and, in some subtypes, mucous membranes and other organs. Currently classified into four main subtypes (EB simplex, junctional EB, dystrophic EB, and Kindler syndrome, mainly based on the level of skin cleavage), the spectrum of EB extends to more than 30 clinical subtypes with pathogenic mutations in at least 18 distinct genes. This review focuses on three recent additions to variants of EB: all are autosomal recessive, and result from mutations in either DST-e (coding for epidermal dystonin, also known as the 230 kDa bullous pemphigoid antigen, BP230), EXPH5 (coding for exophilin-5, also known as Slac2-b), or ITGA3 (coding for the integrin alpha-3 subunit). Each of these new forms of EB is reviewed with respect to the initial gene discovery, clinical features, the current mutation database, and skin pathology. Awareness of these recently described forms of EB is helpful in the clinical evaluation of patients with EB and in defining genotype-phenotype correlation for inherited blistering skin diseases.
Keyword
MeSH Terms
Figure
-
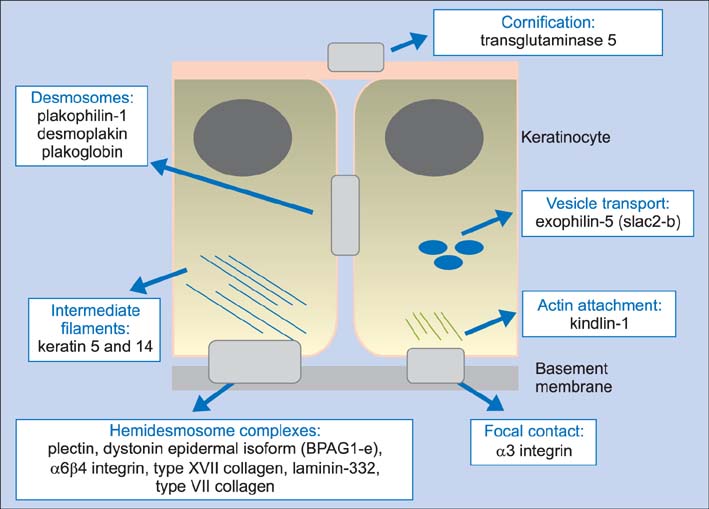
Fig. 1 The molecular basis of epidermolysis bullosa. Mutations in structural components of hemidesmosomes, desmosomes, corneodesmosomes, intermediate filaments, actin microfilaments, focal contacts, and cell vesicle transport underlie a spectrum of skin fragility phenotypes.
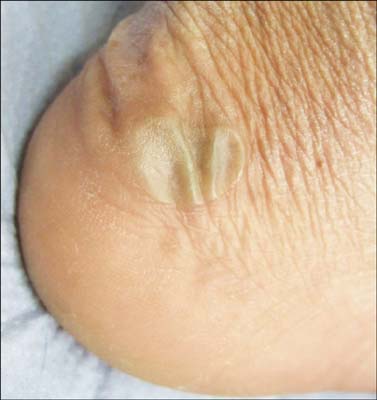
Fig. 2 Clinical features associated with autosomal recessive mutations in DST-e. An intra-epidermal blister on the heel resulting from a homozygous nonsense mutation in DST-e.
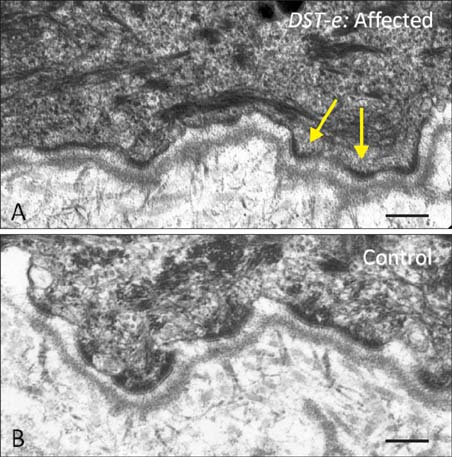
Fig. 3 Transmission electron microscopy of the dermal-epidermal junction in an individual with autosomal recessive mutations in DST-e. (A) Complete lack of hemidesmosomal inner plaques (arrows) in skin from an affected individual. (B) Clearly discernible inner and outer hemidesmosomal plaques are visible in healthy, unaffected skin. Scale bar=0.2 µm.
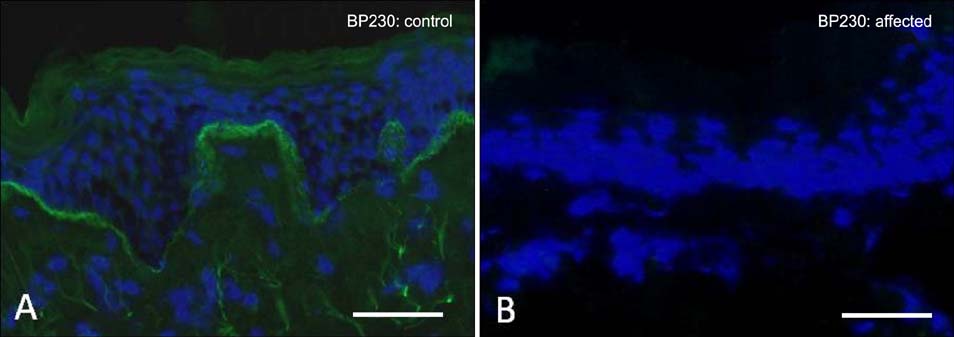
Fig. 4 Immunofluorescence microscopy labeling of skin for DST-e (BP230). (A) In healthy control skin, there is bright linear staining at the dermal-epidermal junction. (B) In contrast, in skin from a patient with biallelic DST-e mutations, there is a complete absence of immunostaining. Scale bar=50 µm.
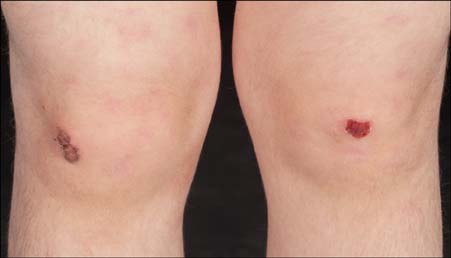
Fig. 5 Clinical features associated with autosomal recessive mutations in EXPH5. There is minor trauma-induced blistering and erosions on both knees, as well as surrounding patchy erythema.
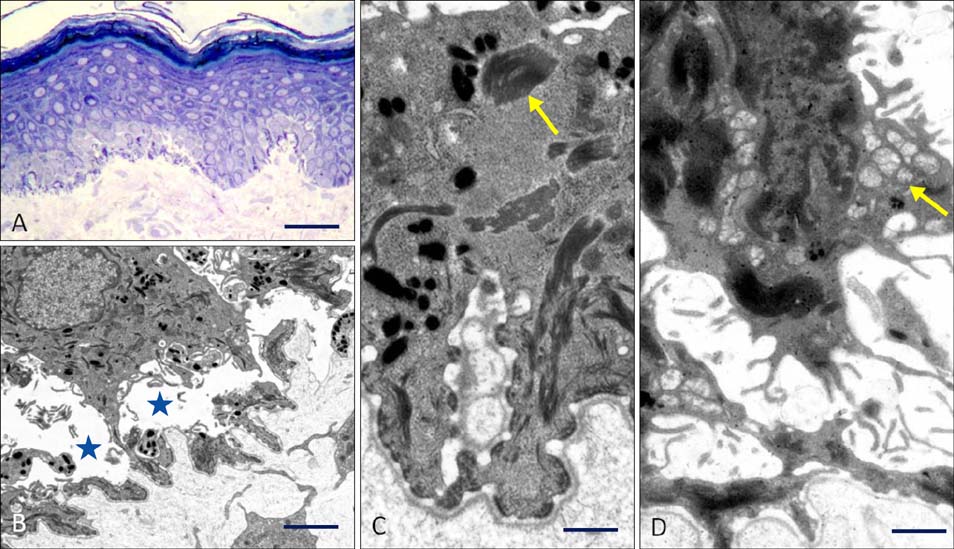
Fig. 6 Skin microscopic abnormalities resulting from mutations in EXPH5. (A) Semi-thin section reveals acanthosis and mild hyperkeratosis. There is also pallor within the basal keratinocyte layer and evidence of darkly stained, condensed cellular material Scale bar=50 µm (Richardson's stain). (B) Transmission electron microscopy confirms a low level, intra-epidermal cleavage (asterisks) between basal keratinocyte nuclei and the cell membrane. Scale bar=5 µm. (C) Higher magnification establishes that the intracellular darkly stained material seen on light microscopy is composed of aggregated keratin tonofilaments (arrow). Scale bar=0.5 µm. (D) Some keratinocytes have numerous perinuclear vesicles (arrow). Scale bar=0.5 µm.
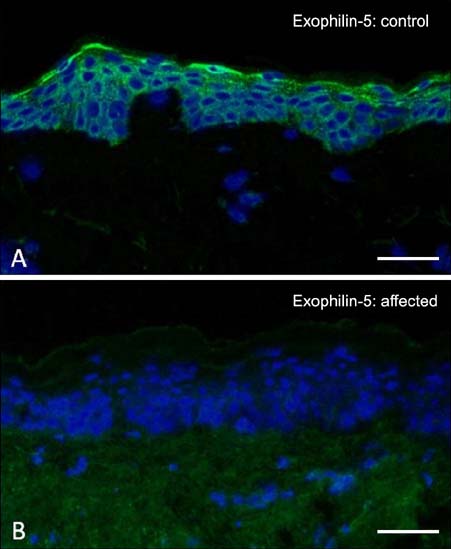
Fig. 7 Immunofluorescence microscopy labeling of skin for exophilin-5. (A) In healthy control skin, there is bright pan-epidermal cytoplasmic staining. (B) In contrast, in skin from a patient with biallelic EXPH5 mutations, there is a complete absence of immunostaining. Scale bar=50 µm.
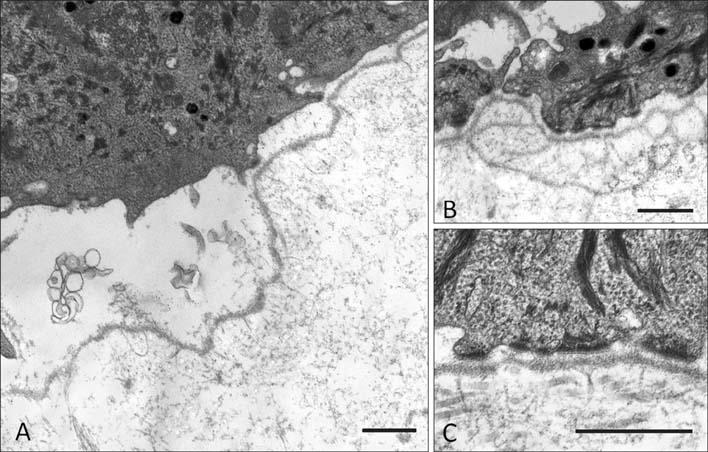
Fig. 8 Transmission electron microscopy of the dermal-epidermal junction in an individual with autosomal recessive mutations in ITGA3. (A) There is cleavage formation within the lamina lucida consistent with a junctional from of epidermolysis bullosa. (B) There is also evidence of focal reduplication of the lamina densa. (C) The ultrastructural appearances of keratin tonofilaments, hemidesmosomes, and anchoring fibrils show no major structural abnormalities. Scale bar=0.5 µm.
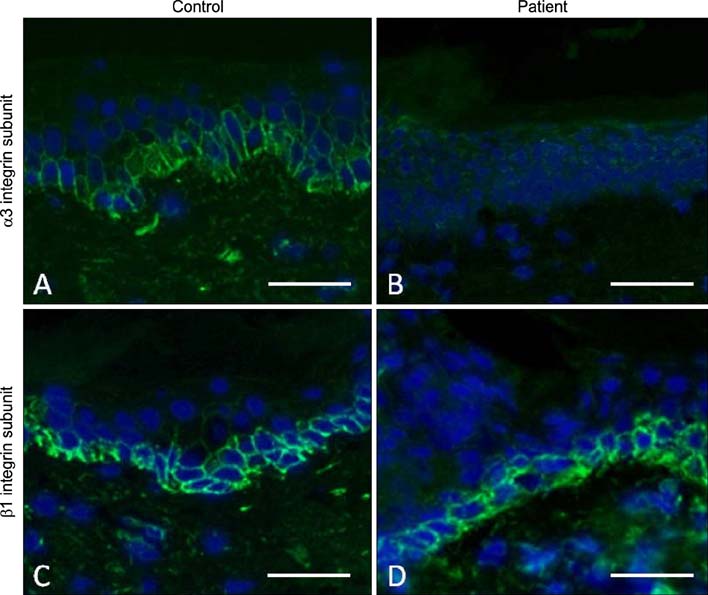
Fig. 9 Immunofluorescence microscopy labeling of skin for the α3 integrin and β1 integrin subunits. (A) In healthy control skin, there is α3 integrin labeling at the cell margins of basal keratinocytes. (B) In contrast, in skin from a patient with biallelic ITGA3 mutations, there is a complete absence of α3 integrin immunostaining. (C) Immunostaining for β1 integrin in healthy control skin shows a similar pattern of labeling to α3 integrin. (D) In skin from a patient with biallelic ITGA3 mutations, no abnormalities in β1 integrin labeling are noted, with the appearances resembling the healthy control skin. Scale bar=50 µm.
Reference
-
1. Salam A, Proudfoot LE, McGrath JA. Inherited blistering skin diseases: underlying molecular mechanisms and emerging therapies. Ann Med. 2014; 46:49–61.
Article2. Fine JD, Bruckner-Tuderman L, Eady RA, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014; 70:1103–1126.
Article3. Has C, Bruckner-Tuderman L. The genetics of skin fragility. Annu Rev Genomics Hum Genet. 2014; 15:245–268.
Article4. Groves RW, Liu L, Dopping-Hepenstal PJ, Markus HS, Lovell PA, Ozoemena L, et al. A homozygous nonsense mutation within the dystonin gene coding for the coiled-coil domain of the epithelial isoform of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J Invest Dermatol. 2010; 130:1551–1557.
Article5. Liu L, Dopping-Hepenstal PJ, Lovell PA, Michael M, Horn H, Fong K, et al. Autosomal recessive epidermolysis bullosa simplex due to loss of BPAG1-e expression. J Invest Dermatol. 2012; 132:742–744.
Article6. Takeichi T, Nanda A, Liu L, Aristodemou S, McMillan JR, Sugiura K, et al. Founder mutation in dystonin-e underlying autosomal recessive epidermolysis bullosa simplex in Kuwait. Br J Dermatol. 2015; 172:527–531.
Article7. Michael M, Begum R, Fong K, Pourreyron C, South AP, McGrath JA, et al. BPAG1-e restricts keratinocyte migration through control of adhesion stability. J Invest Dermatol. 2014; 134:773–782.
Article8. McGrath JA, Stone KL, Begum R, Simpson MA, Dopping-Hepenstal PJ, Liu L, et al. Germline Mutation in EXPH5 Implicates the Rab27B Effector Protein Slac2-b in Inherited Skin Fragility. Am J Hum Genet. 2012; 91:1115–1121.
Article9. Fukuda M. Rab27 effectors, pleiotropic regulators in secretory pathways. Traffic. 2013; 14:949–963.
Article10. Pigors M, Schwieger-Briel A, Leppert J, Kiritsi D, Kohlhase J, Bruckner-Tuderman L, et al. Molecular heterogeneity of epidermolysis bullosa simplex: contribution of EXPH5 mutations. J Invest Dermatol. 2014; 134:842–845.
Article11. Liu L, Mellerio JE, Martinez AE, McMillan JR, Aristodemou S, Parsons M, et al. Mutations in EXPH5 result in autosomal recessive inherited skin fragility. Br J Dermatol. 2014; 170:196–199.
Article12. Rashidghamat E, Ozoemena L, Liu L, McGrath JA, Martinez AE, Mellerio JE. Mutations in EXPH5 (exophilin-5) underlie a rare subtype of autosomal recessive epidermolysis bullosa simplex. Br J Dermatol. 2015; DOI: 10.1111/bjd.14047. [Epub ahead of print].13. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009; 10:513–525.
Article14. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010; 12:19–30.
Article15. Has C, Spartà G, Kiritsi D, Weibel L, Moeller A, Vega-Warner V, et al. Integrin α3 mutations with kidney, lung, and skin disease. N Engl J Med. 2012; 366:1508–1514.
Article16. Nicolaou N, Margadant C, Kevelam SH, Lilien MR, Oosterveld MJ, Kreft M, et al. Gain of glycosylation in integrin α 3 causes lung disease and nephrotic syndrome. J Clin Invest. 2012; 122:4375–4387.
Article17. Yalcin EG, He Y, Orhan D, Pazzagli C, Emiralioglu N, Has C. Crucial role of posttranslational modifications of integrin α3 in interstitial lung disease and nephrotic syndrome. Hum Mol Genet. 2015; 24:3679–3688.
Article18. Watt FM. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002; 21:3919–3926.
Article19. Hodivala-Dilke KM, DiPersio CM, Kreidberg JA, Hynes RO. Novel roles for alpha3beta1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J Cell Biol. 1998; 142:1357–1369.
Article20. Margadant C, Charafeddine RA, Sonnenberg A. Unique and redundant functions of integrins in the epidermis. FASEB J. 2010; 24:4133–4152.
Article21. Shukrun R, Vivante A, Pleniceanu O, Vax E, Anikster Y, Dekel B, et al. A human integrin-α3 mutation confers major renal developmental defects. PLoS One. 2014; 9:e90879.
Article22. Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, et al. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996; 122:3537–3547.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Localized Epidermolysis Bullosa Acquisita Limited to the Face
- Epidermolysis Bullosa Acquisita: A Complete Remissions versus Patients with Long-term Persistent Activities
- A Case of Recessive Epidermolysis Bullosa Dystrophica
- A Case of Epidermolysis Bullosa Dystrophica
- A Case of Epidermolysis Bullosa Letalis
