Single Nucleotide Deletion Mutation of KCNH2 Gene is Responsible for LQT Syndrome in a 3-Generation Korean Family
- Affiliations
-
- 1Department of Biomedical Sciences, Seoul St. Mary's Hospital, The Catholic University of Korea, Seoul, Korea. sjkyoon@catholic.ac.kr
- 2Division of Cardiovascular Medicine, Seoul St. Mary's Hospital, The Catholic University of Korea, Seoul, Korea.
- KMID: 1793058
- DOI: http://doi.org/10.3346/jkms.2013.28.9.1388
Abstract
- Long QT syndrome (LQTS) is characterized by the prolongation of the QT interval in ECG and manifests predisposition to life threatening arrhythmia which often leads to sudden cardiac death. We encountered a 3-generation family with 5 affected family members in which LQTS was inherited in autosomal dominant manner. The LQTS is considered an ion channel disorder in which the type and location of the genetic mutation determines to a large extent the expression of the clinical syndrome. Upon screening of the genomic sequences of cardiac potassium ion channel genes, we found a single nucleotide C deletion mutation in the exon 3 of KCNH2 gene that co-segregates with the LQTS in this family. This mutation presumably resulted in a frameshift mutation, P151fs+15X. This study added a new genetic cause to the pool of mutations that lead to defected potassium ion channels in the heart.
MeSH Terms
-
Adolescent
Adult
Aged
Aged, 80 and over
Asian Continental Ancestry Group/*genetics
DNA Mutational Analysis
Ether-A-Go-Go Potassium Channels/*genetics
Exons
Female
Frameshift Mutation
Genotype
Humans
Long QT Syndrome/*diagnosis/genetics
Male
Middle Aged
Pedigree
Republic of Korea
Sequence Deletion
Ether-A-Go-Go Potassium Channels
Figure
-

Fig. 1 It showed typical figures of 'Torsades de Pointes', In (A), the black arrows indicate long QT interval and T wave alternans.
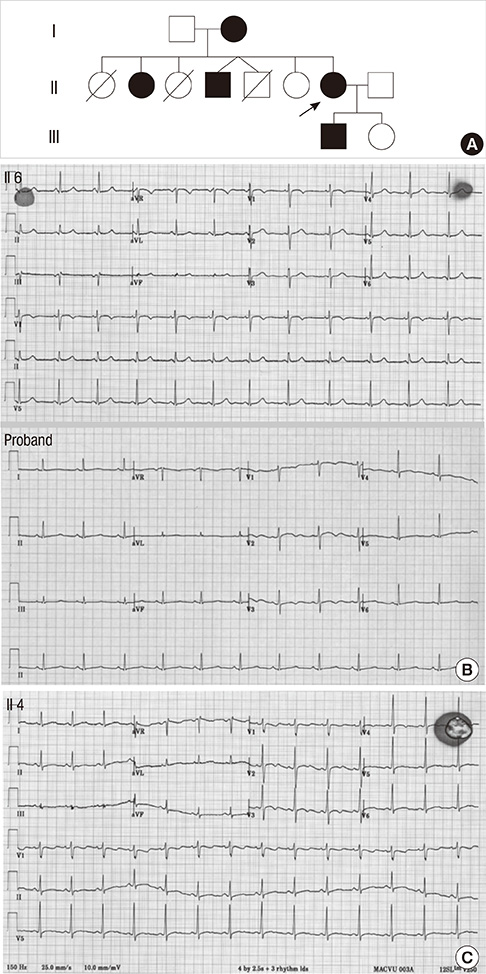
Fig. 2 Pedigree and Electrocardiogram. (A) Pedigree of the LQTS family; filled symbols represent affected members whereas empty symbols normal members. Arrow indicates the proband. (B) Electrocardiogram of normal (II6) and the proband (II7). (C) Electrocardiogram of II4 displays atypically short QTc interval than those of the other family members. This patient also contains del453 C mutation (151Pfs+15X) in the KCNH2 gene.
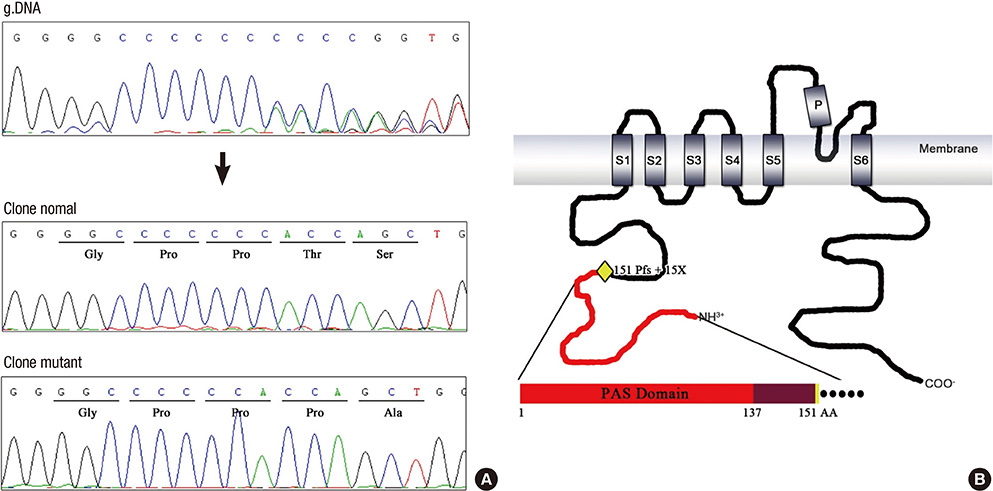
Fig. 3 Chromatogram of 453delC mutation in the KCNH2 gene (A) and schematic representation of the KCNH2 protein (B). Yellow diamond depicts the mutation (151Pfs+15X) described in this study.
Reference
-
1. Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005; 115:2018–2024.2. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, et al. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000; 102:1178–1185.3. Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA, Christiansen M. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat. 2009; 30:1486–1511.4. Itoh H, Shimizu W, Hayashi K, Yamagata K, Sakaguchi T, Ohno S, Makiyama T, Akao M, Ai T, Noda T, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart Rhythm. 2010; 7:1411–1418.5. Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957; 54:59–68.6. Sagie A, Larson MG, Goldberg RJ, Bengtson JR, Levy D. An improved method for adjusting the QT interval for heart rate (the Framingham Heart Study). Am J Cardiol. 1992; 70:797–801.7. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993; 88:782–784.8. Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, Hall WJ, Weitkamp L, Vincent GM, Garson A Jr, et al. The long QT syndrome: prospective longitudinal study of 328 families. Circulation. 1991; 84:1136–1144.9. Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, Benhorin J, Locati EH, Towbin JA, Keating MT, et al. Influence of genotype on the clinical course of the long-QT syndrome: International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998; 339:960–965.10. Moss AJ, Robinson JL, Gessman L, Gillespie R, Zareba W, Schwartz PJ, Vincent GM, Benhorin J, Heilbron EL, Towbin JA, et al. Comparison of clinical and genetic variables of cardiac events associated with loud noise versus swimming among subjects with the long QT syndrome. Am J Cardiol. 1999; 84:876–879.11. Ali RH, Zareba W, Moss AJ, Schwartz PJ, Benhorin J, Vincent GM, Locati EH, Priori S, Napolitano C, Towbin JA, et al. Clinical and genetic variables associated with acute arousal and nonarousal-related cardiac events among subjects with long QT syndrome. Am J Cardiol. 2000; 85:457–461.12. Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998; 95:649–655.13. Clancy CE, Rudy Y. Cellular consequences of HERG mutations in the long QT syndrome: precursors to sudden cardiac death. Cardiovasc Res. 2001; 50:301–313.14. Thomas D, Kiehn J, Katus HA, Karle CA. Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing. Cardiovasc Res. 2003; 60:235–241.15. Laitinen P, Fodstad H, Piippo K, Swan H, Toivonen L, Viitasalo M, Kaprio J, Kontula K. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Hum Mutat. 2000; 15:580–581.16. Fodstad H, Swan H, Laitinen P, Piippo K, Paavonen K, Viitasalo M, Toivonen L, Kontula K. Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann Med. 2004; 36:53–63.17. Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem. 1999; 274:10113–10118.18. Wang J, Myers CD, Robertson GA. Dynamic control of deactivation gating by a soluble amino-terminal domain in HERG K(+) channels. J Gen Physiol. 2000; 115:749–758.19. Li X, Xu J, Li M. The human delta1261 mutation of the HERG potassium channel results in a truncated protein that contains a subunit interaction domain and decreases the channel expression. J Biol Chem. 1997; 272:705–708.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A novel KCNH2 frameshift mutation (c.46delG) associated with high risk of sudden death in a family with congenital long QT syndrome type 2
- Proarrhythmogenic Effect of the L532P and N588K KCNH2 Mutations in the Human Heart Using a 3D Electrophysiological Model
- Novel insertion mutation of ABCB1 gene in an ivermectin-sensitive Border Collie
- Novel Point Mutation of EBSS Gene Coexisted with 1p36 Deletion
- Mutations and Polymorphims of MECP2 Gene in Rett Syndrome
