A rare pathogenic variant identified in a heart transplant recipient with hereditary transthyretin amyloidosis: a case report
- Affiliations
-
- 1Division of Cardiology, Department of Internal Medicine, Cardiovascular Center, Incheon Sejong Hospital, Incheon, Korea
- KMID: 2548349
- DOI: http://doi.org/10.12701/jyms.2023.00241
Abstract
- Hereditary transthyretin (ATTRv) amyloidosis is a rare and complex genetic disorder that can lead to life-threatening cardiac amyloidosis and rapid disease progression. Early diagnosis and treatment with disease-modifying drugs can improve patient outcomes; however, heart transplantation may be necessary in some patients. We present the unique case of a 65-year-old Korean woman diagnosed with ATTRv amyloidosis after experiencing progressive neurological symptoms, followed by heart failure. Despite the absence of significant symptoms of heart failure, subsequent screening revealed cardiac amyloid infiltration, which caused left ventricular hypertrophy and rapid disease progression. The patient underwent successful heart transplantation, and subsequent genetic testing revealed a pathogenic variant, NM_000371.3:c.425T>C (p.Val142Ala), which affects both the nerves and heart and has not been previously reported in Korea. Our report underscores the potential benefits of heart transplantation in managing advanced ATTRv amyloidosis and emphasizes the need for continued research on the genetic heterogeneity of the disease. Clinicians should consider ATTRv amyloidosis in the differential diagnosis of patients presenting with neurological symptoms and heart failure, particularly in those with a family history of the disease.
Keyword
Figure
-
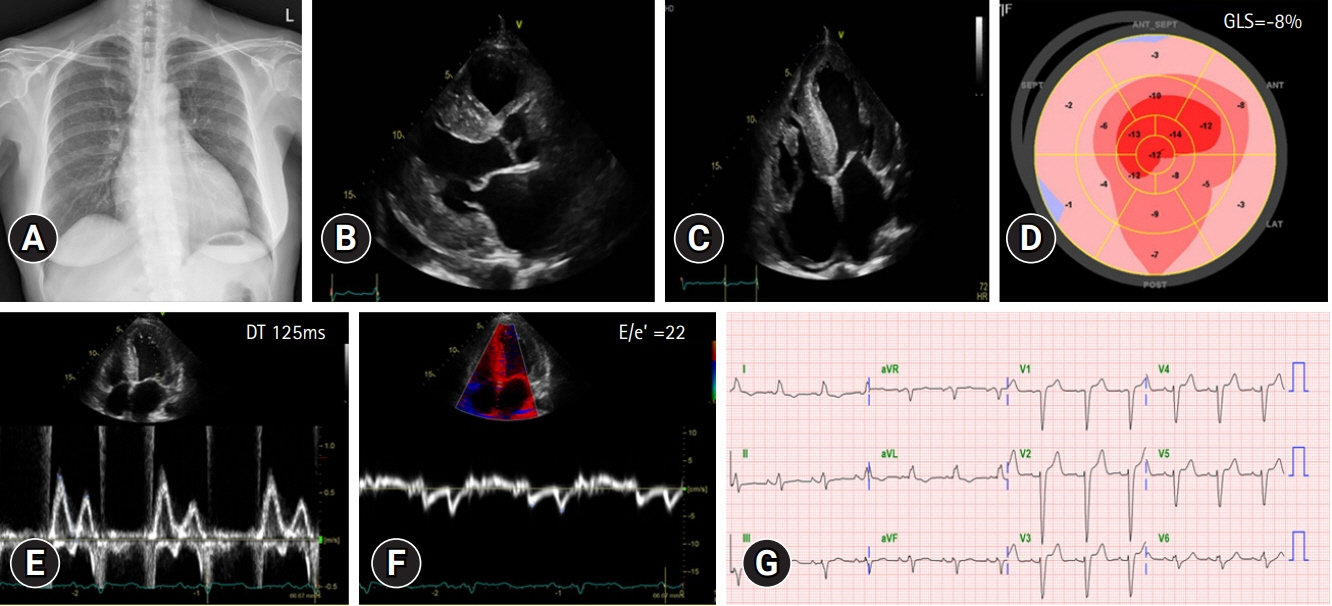
Fig. 1. (A) Initial chest X-ray shows cardiomegaly. (B) Transthoracic echocardiography (TTE) images. Parasternal long axis view. The images show concentric hypertrophy of the left ventricular wall and a small amount of pericardial effusion. (C) Apical four-chamber view of TTE. (D) Global longitudinal strain (GLS) shows reduced longitudinal strain with apical sparing pattern. (E, F) Mitral inflow and mitral annular velocity show grade III diastolic dysfunction. (G) Initial electrocardiography. DT, deceleration time.

Fig. 2. Results of the nerve conduction study of the patient. The sensory nerve study demonstrates increased nerve latency and decreased amplitude and peak velocity of the median, sural, and superficial peroneal nerves. The motor nerve study demonstrates decreased amplitude in both peroneal nerves. ADM, abductor digiti minimi; AH, abductor hallucis; Amp, amplitude; APB, abductor pollicis brevis; EDB, extensor digitorum brevis; L, left; Lat, latency; MNC, motor nerve conduction; R, right; SNC, sensory nerve conduction; Vel, velocity; Rec, recommendation; PP, peak to peak; Dur, duration.

Fig. 3. Technetium-99m-labeled 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. Intense cardiac uptake (arrow) of the radiotracer is observed in both ventricles. (A) Axial view and (B) anterior view. DPD, 3-diphosphono-1,2-propanodicarboxylic acid.

Fig. 4. Microscopic findings of endomyocardial biopsy. (A) The specimen shows positivity for transthyretin (immunohistochemical stain for transthyretin). (B) The Congo Red stain shows pink-red deposits of amyloid (Congo Red stain).
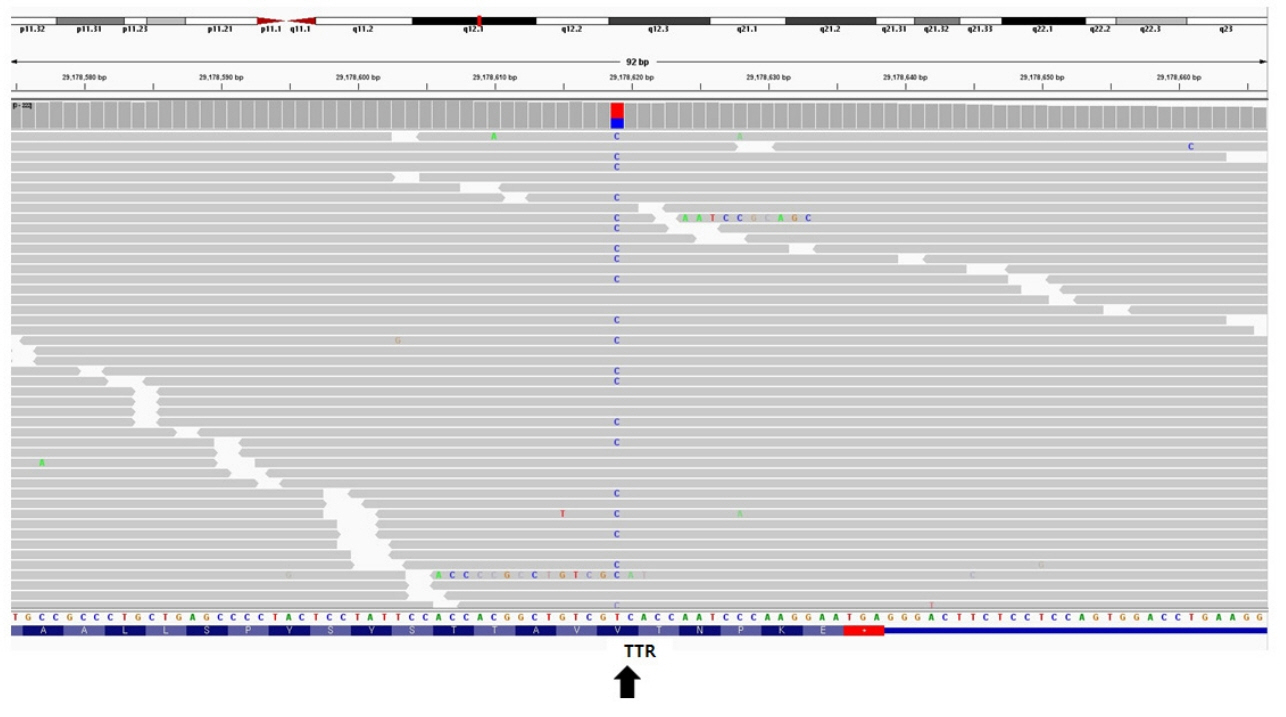
Fig. 5. Integrated genomics viewer snapshot of the transthyretin variant (NM_000371.3:c.425T>C) identified in the patient (arrow). TTR, transthyretin.
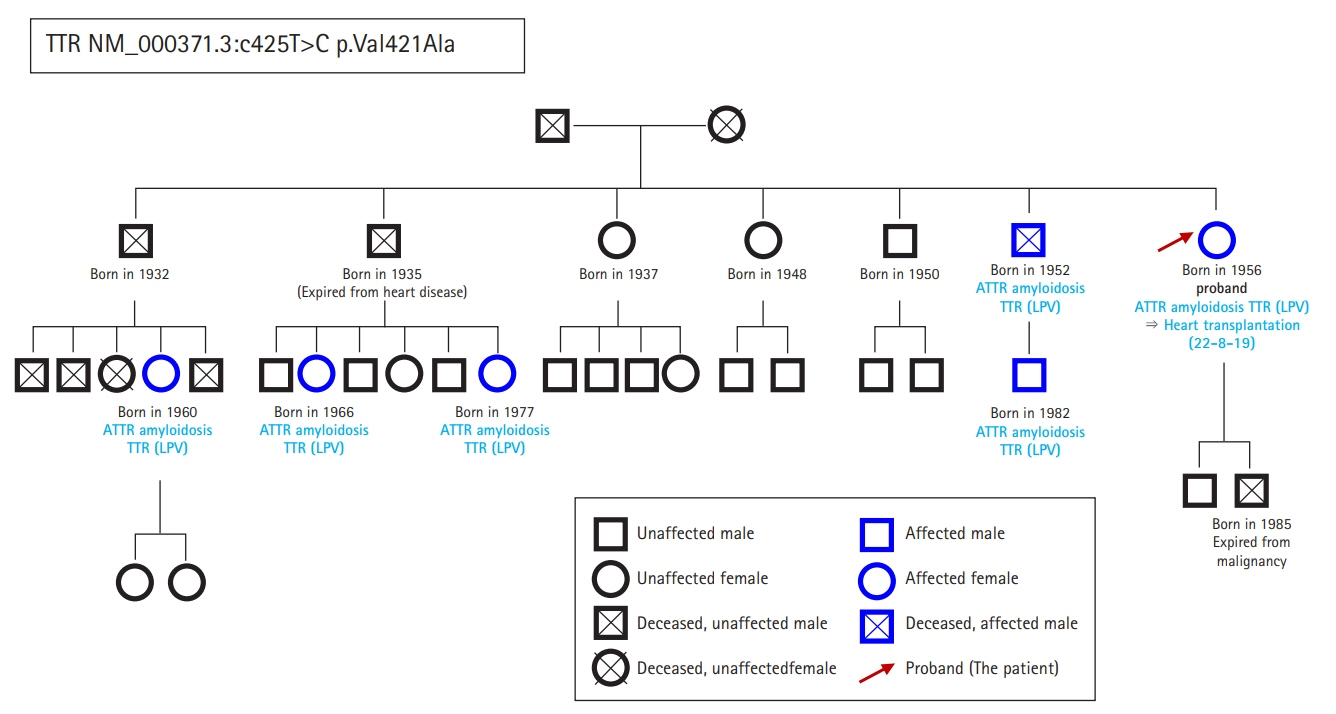
Fig. 6. Patient pedigree. The squares and circles represent males and females, respectively, and X marks indicate deceased family members. Blue represents possession of the character under study and black represents an absence. ATTR, amyloid transthyretin; LPV, likely pathogenic variant; TTR, transthyretin.
Reference
-
References
1. Damy T, Kristen AV, Suhr OB, Maurer MS, Planté-Bordeneuve V, Yu CR, et al. Transthyretin cardiac amyloidosis in continental Western Europe: an insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur Heart J. 2019; 43:391–400.
Article2. Rapezzi C, Aimo A, Serenelli M, Barison A, Vergaro G, Passino C, et al. Critical comparison of documents from scientific societies on cardiac amyloidosis: JACC State-of-the-Art Review. J Am Coll Cardiol. 2022; 79:1288–303.3. Lioncino M, Monda E, Palmiero G, Caiazza M, Vetrano E, Rubino M, et al. Cardiovascular involvement in transthyretin cardiac amyloidosis. Heart Fail Clin. 2022; 18:73–87.
Article4. Griffin JM, Rosenthal JL, Grodin JL, Maurer MS, Grogan M, Cheng RK. ATTR amyloidosis: current and emerging management strategies: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2021; 3:488–505.5. Coelho T, Conceição I, Waddington-Cruz M, Keohane D, Sultan MB, Chapman D, et al. A natural history analysis of asymptomatic TTR gene carriers as they develop symptomatic transthyretin amyloidosis in the Transthyretin Amyloidosis Outcomes Survey (THAOS). Amyloid. 2022; 29:228–36.
Article6. Ghoneem A, Bhatti AW, Khadke S, Mitchell J, Liu J, Zhang K, et al. Real-world efficacy of tafamidis in patients with transthyretin amyloidosis and heart failure. Curr Probl Cardiol. 2023; 48:101667.
Article7. Dispenzieri A, Coelho T, Conceição I, Waddington-Cruz M, Wixner J, Kristen AV, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis. 2022; 17:236.8. Chandrashekar P, Alhuneafat L, Mannello M, Al-Rashdan L, Kim MM, Dungu J, et al. Prevalence and outcomes of p.Val142Ile TTR amyloidosis cardiomyopathy: a systematic review. Circ Genom Precis Med. 2021; 14:e003356.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Hereditary Transthyretin Amyloidosis Misdiagnosed as Demyelinating Neuropathy: A Report of Three Cases
- Transthyretin Cardiac Amyloidosis: A Case Report
- Familial Amyloidotic Polyneuropathy With Transthyretin Gene Mutation
- Tafamidis for Cardiac Transthyretin Amyloidosis
- Familial Transthyretin Amyloidosis with Variant Asp38Ala Presenting with Orthostatic Hypotension and Chronic Diarrhea
