Tubulopathy: the clinical and genetic approach in diagnosis
- Affiliations
-
- 1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Republic of Korea
- KMID: 2544232
- DOI: http://doi.org/10.3339/ckd.23.001
Abstract
- Remarkable advances in genetic diagnosis expanded our knowledge about inherited tubulopathies and other genetic kidney diseases. This review suggests a simple categorization of inherited tubular disease, clarifies the concept of autosomal dominant tubulointerstitial kidney disease (ADTKD), and introduces novel therapies developed for tubulopathies. Facing patients with suspicious tubular disorders, clinicians should first evaluate the status of volume and acid-base. This step helps the clinicians to localize the affected segment and to confirm genetic diagnosis. ADTKD is a recently characterized disease entity involving tubules. The known causative genes are UMOD, MUC1, REN, and HNF1β. Still, only half of ADTKD patients show mutations for these four identified genes. Whole exome sequencing is a suitable diagnostic tool for tubulopathies, especially for ADTKD. Genetic approaches to treat tubulopathies have progressed recently. Despite the practical obstacles, novel therapies targeting inherited tubulopathies are currently in development.
Figure
-
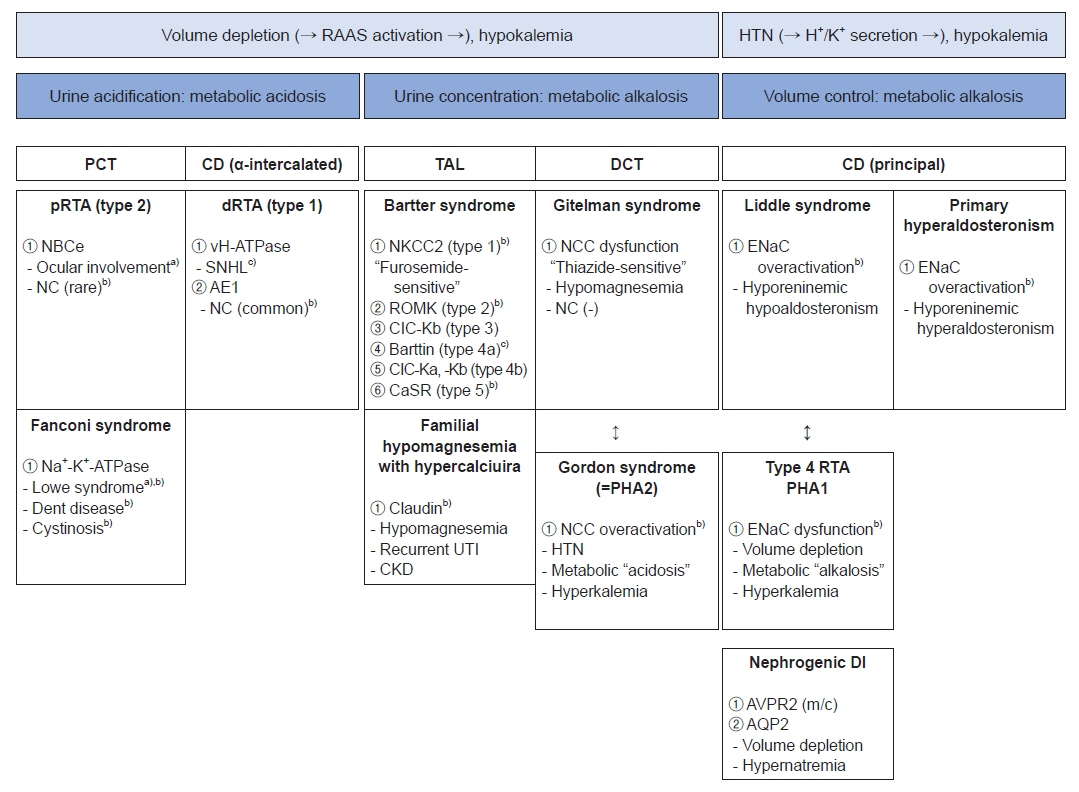
Fig. 1. Clinical approach to inherited tubulopathies. RAAS, renin-angiotensin-aldosterone system; HTN, hypertension; PCT, proximal convoluted tubule; CD, collecting duct; TAL, thick ascending limb of Henle’s loop; DCT, distal convoluted tubule; pRTA, proximal renal tubular acidosis; dRTA, distal renal tubular acidosis; NBCe, electrogenic sodium bicarbonate cotransporter 1; NC, nephrocalcinosis; vH-ATPase, vacuolar type (H+)-ATPase; SNHL, sensorineural hearing loss; AE1, anion exchanger 1; NKCC2, Na+-K+-2Cl– cotransporter; ROMK, renal outer medullary potassium; CIC-Kb, chloride channel-Kb; CIC-Ka, chloride channel-Ka; CaSR, calcium sensing receptor; UTI, urinary tract infection; CKD, chronic kidney disease; NCC, Na+-Cl– cotransporter; PHA1, pseudohypoaldosteronism (PHA) type 1; PHA2, PHA type 2; ENaC, epithelial sodium channel; RTA, renal tubular acidosis; DI, diabetes insipidus; AVPR2, arginine vasopressin receptor 2; AQP2, aquaporin 2. a)Tubulopathy with ocular involvement. b)Tubulopathy with nephrocalcinosis. c)Tubulopathy with sensorineural hearing loss.
Reference
-
References
1. Downie ML, Lopez Garcia SC, Kleta R, Bockenhauer D. Inherited tubulopathies of the kidney: insights from genetics. Clin J Am Soc Nephrol. 2021; 16:620–30.2. Kleta R, Bockenhauer D. Salt-losing tubulopathies in children: what’s new, what's controversial? J Am Soc Nephrol. 2018; 29:727–39.
Article3. Kermond R, Mallett A, McCarthy H. A clinical approach to tubulopathies in children and young adults. Pediatr Nephrol. 2023; 38:651–62.
Article4. Foreman JW. Fanconi syndrome. Pediatr Clin North Am. 2019; 66:159–67.
Article5. Walsh SB, Unwin RJ. Renal tubular disorders. Clin Med (Lond). 2012; 12:476–9.
Article6. Raina R, Krishnappa V, Das A, Amin H, Radhakrishnan Y, Nair NR, et al. Overview of monogenic or mendelian forms of hypertension. Front Pediatr. 2019; 7:263.
Article7. Alexander RT, Cordat E, Chambrey R, Dimke H, Eladari D. Acidosis and urinary calcium excretion: insights from genetic disorders. J Am Soc Nephrol. 2016; 27:3511–20.
Article8. Alexander RT, Bitzan M. Renal tubular acidosis. Pediatr Clin North Am. 2019; 66:135–57.
Article9. Klootwijk ED, Reichold M, Unwin RJ, Kleta R, Warth R, Bockenhauer D. Renal Fanconi syndrome: taking a proximal look at the nephron. Nephrol Dial Transplant. 2015; 30:1456–60.
Article10. Bokenkamp A, Ludwig M. The oculocerebrorenal syndrome of Lowe: an update. Pediatr Nephrol. 2016; 31:2201–12.
Article11. De Matteis MA, Staiano L, Emma F, Devuyst O. The 5-phosphatase OCRL in Lowe syndrome and Dent disease 2. Nat Rev Nephrol. 2017; 13:455–70.12. Blaine J, Chonchol M, Levi M. Renal control of calcium, phosphate, and magnesium homeostasis. Clin J Am Soc Nephrol. 2015; 10:1257–72.
Article13. Besouw MT, Kleta R, Bockenhauer D. Bartter and Gitelman syndromes: questions of class. Pediatr Nephrol. 2020; 35:1815–24.
Article14. Walsh PR, Tse Y, Ashton E, Iancu D, Jenkins L, Bienias M, et al. Clinical and diagnostic features of Bartter and Gitelman syndromes. Clin Kidney J. 2018; 11:302–9.
Article15. Claverie-Martin F. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis: clinical and molecular characteristics. Clin Kidney J. 2015; 8:656–64.
Article16. Bockenhauer D, Bichet DG. Pathophysiology, diagnosis and management of nephrogenic diabetes insipidus. Nat Rev Nephrol. 2015; 11:576–88.
Article17. Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, et al. Autosomal dominant tubulointerstitial kidney disease: diagnosis, classification, and management: a KDIGO consensus report. Kidney Int. 2015; 88:676–83.18. Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, et al. Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers. 2019; 5:60.
Article19. Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019; 380:142–51.
Article20. Bleyer AJ, Wolf MT, Kidd KO, Zivna M, Kmoch S. Autosomal dominant tubulointerstitial kidney disease: more than just HNF1β. Pediatr Nephrol. 2022; 37:933–46.
Article21. Sinha R, Pradhan S, Banerjee S, Jahan A, Akhtar S, Pahari A, et al. Whole-exome sequencing and variant spectrum in children with suspected inherited renal tubular disorder: the East India Tubulopathy Gene Study. Pediatr Nephrol. 2022; 37:1811–36.
Article22. Ashton EJ, Legrand A, Benoit V, Roncelin I, Venisse A, Zennaro MC, et al. Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int. 2018; 93:961–7.
Article23. Hildebrandt F. Genetic kidney diseases. Lancet. 2010; 375:1287–95.
Article24. Rubin JD, Barry MA. Improving molecular therapy in the kidney. Mol Diagn Ther. 2020; 24:375–96.
Article25. Chung DC, Fogelgren B, Park KM, Heidenberg J, Zuo X, Huang L, et al. Adeno-associated virus-mediated gene transfer to renal tubule cells via a retrograde ureteral approach. Nephron Extra. 2011; 1:217–23.
Article26. Cherqui S. Hematopoietic stem cell gene therapy for cystinosis: from bench-to-bedside. Cells. 2021; 10:3273.
Article27. Cystinosis Research Foundation (CRF) News. Stem Cell Trial Phase I/II Updates by Stephanie Cherqui, PhD [Internet]. CRF;2022. [cited 2022 Dec 10]. Available from: https://www.cystinosisresearch.org/stem-cell-clinical-trial-update-by-stephanie-cherqui-phd/.28. Berquez M, Gadsby JR, Festa BP, Butler R, Jackson SP, Berno V, et al. The phosphoinositide 3-kinase inhibitor alpelisib restores actin organization and improves proximal tubule dysfunction in vitro and in a mouse model of Lowe syndrome and Dent disease. Kidney Int. 2020; 98:883–96.
