Child Kidney Dis.
2021 Dec;25(2):128-132. 10.3339/jkspn.2021.25.2.128.
Morning Glory Syndrome associated with Autosomal Dominant Alport Syndrome with a Heterozygous COL4A4 Mutation
- Affiliations
-
- 1Department of Pediatrics, Busan Paik Hospital, Inje University, Busan, Korea
- 2Department of Ophthalmology, Busan Paik Hospital, Inje University, Busan, Korea
- 3Department of Patholgy, Busan Paik Hospital, Inje University, Busan, Korea
- 4Busan Regional Rare Disease Center, Busan, Korea
- 53billion, Inc., Seoul, Korea
- 6Department of Laboratory Medicine, Yangsan Busan National University Hospital, Yangsan, Korea
- KMID: 2524515
- DOI: http://doi.org/10.3339/jkspn.2021.25.2.128
Abstract
- Morning glory syndrome (MGS) is a rare congenital optic disc anomaly with a characteristic fundal finding with severe visual impairment. It may occur in association with various systemic manifestations, even though most of the reported cases were isolated. A 6-year-old male visited the nephrology clinic with a history of microscopic hematuria and at the age of 12 years, he was diagnosed thin glomerular basement membrane nephropathy by kidney biopsy. After the following years, the patient had progressive deterioration of visual acuity, and diagnosed as MGS. Whole Exome Sequencing of this patient and his mother revealed heterozygous COL4A4 mutations [c.81_86del (p.Ile29_Leu30del)]. It is more reasonable to consider MGS seen in this patient as a coincidental finding of autosomal dominant Alport syndrome. To our knowledge, this case represents the first case report of autosomal dominant Alport syndrome associated with MGS.
Keyword
Figure
-
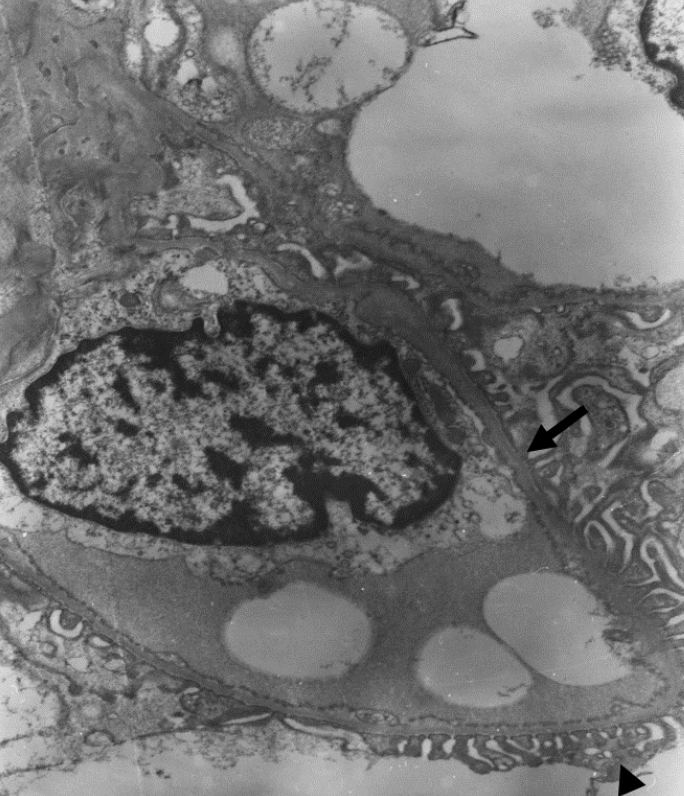
Fig. 1. Electron microscopic examination of glomerulus. Glomerular basement membrane (GBM) (arrow) was diffusely thinned (166.7–208.3 nm) without foot process effacement (arrow head) in electron microscopic examination (x5,000).
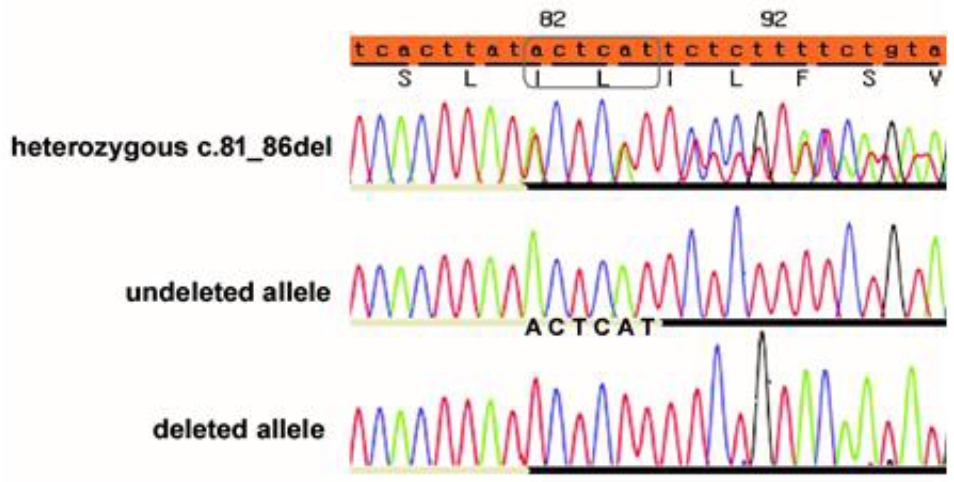
Fig. 2. Electropherograms of the COL4A4 gene. Sequence analysis revealed a heterozygous ACTCAT deletion between cDNA position 81 and 86. In the forward strand (top), heterozygous peaks from the deletion site (81–86) represent a mixture of the undeleted (middle) and deleted (bottom) alleles.

Fig. 3. Fundoscopic examination on both eyes. Enlarged disc with white center (arrow) is shown in the left eye resulting in similar look of petals of a flower.
Reference
-
References
1. Piel CF, Biava CG, Goodman JR. Glomerular basement membrane attenuation in familial nephritis and "benign" hematuria. J Pediatr. 1982; 101:358–65.
Article2. Wang YY, Rana K, Tonna S, Lin T, Sin L, Savige J. COL4A3 mutations and their clinical consequences in thin basement membrane nephropathy (TBMN). Kidney Int. 2004; 65:786–90.3. Buzza M, Wilson D, Savige J. Segregation of hematuria in thin basement membrane disease with haplotypes at the loci for Alport syndrome. Kidney Int. 2001; 59:1670–6.
Article4. Piccini M, Casari G, Zhou J, Bruttini M, Volti SL, Ballabio A, et al. Evidence for genetic heterogeneity in benign familial hematuria. Am J Nephrol. 1999; 19:464–7.
Article5. Voskarides K, Patsias C, Pierides A, Deltas C. COL4A3 founder mutations in Greek-Cypriot families with thin basement membrane nephropathy and focal segmental glomerulosclerosis dating from around 18th century. Genet Test. 2008; 12:273–8.6. Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol. 2015; 10:703–9.
Article7. Imafuku A, Nozu K, Sawa N, Nakanishi K, Ubara Y. How to resolve confusion in the clinical setting for the diagnosis of heterozygous COL4A3 or COL4A4 gene variants? Discussion and suggestions from nephrologists. Clin Exp Nephrol. 2020; 24:651–6.
Article8. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV α345: a position paper of the Alport syndrome classification working group. Kidney Int. 2018; 93:1045–51.
Article9. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A4 variant? Kidney Int Rep. 2018; 3:1239–41.10. Kindler P. Morning glory syndrome: unusual congenital optic disk anomaly. Am J Ophthalmol. 1970; 69:376–84.
Article11. King AM, Bowen DI, Goulding P, Doran RM. Aicardi syndrome. Br J Ophthalmol. 1998; 82:457.
Article12. Taskintuna I, Oz O, Teke MY, Kocak H, Firat E. Morning glory syndrome: association with moyamoya disease, midline cranial defects, central nervous system anomalies, and persistent hyaloid artery remnant. Retina. 2003; 23:400–2.13. Imafuku A, Nozu K, Sawa N, Hasegawa E, Hiramatsu R, Kawada M, et al. Autosomal dominant form of type IV collagen nephropathy exists among patients with hereditary nephritis difficult to diagnose clinicopathologically. Nephrology (Carlton). 2018; 23:940–7.14. Helen S, Judy S, Vanessa S, Stephen A, Frances A. COL4A3/COL4A4 Mutations and Features in Individuals with Autosomal Recessive Alport Syndrome. J Am Soc Nephrol. 24:2013; 1945–54.
