Steroidogenic electron-transfer factors and their diseases
- Affiliations
-
- 1Department of Pediatrics, Center for Reproductive Sciences and Institute for Human Genetics, University of California, San Francisco, CA, USA
- KMID: 2520506
- DOI: http://doi.org/10.6065/apem.2142154.077
Abstract
- Most steroidogenesis disorders are caused by mutations in genes encoding the steroidogenic enzymes, but work in the past 20 years has identified related disorders caused by mutations in the genes encoding the cofactors that transport electrons from NADPH to P450 enzymes. Most P450s are microsomal and require electron donation by P450 oxidoreductase (POR); by contrast, mitochondrial P450s require electron donation via ferredoxin reductase (FdxR) and ferredoxin (Fdx). POR deficiency is the most common and best-described of these new forms of congenital adrenal hyperplasia. Severe POR deficiency is characterized by the Antley-Bixler skeletal malformation syndrome and genital ambiguity in both sexes, and hence is easily recognized, but mild forms may present only with infertility and subtle disorders of steroidogenesis. The common POR polymorphism A503V reduces catalysis by P450c17 (17-hydroxylase/17,20-lyase) and the principal drugmetabolizing P450 enzymes. The 17,20-lyase activity of P450c17 requires the allosteric action of cytochrome b5, which promotes interaction of P450c17 with POR, with consequent electron transfer. Rare b5 mutations are one of several causes of 17,20-lyase deficiency. In addition to their roles with steroidogenic mitochondrial P450s, Fdx and FdxR participate in the synthesis of iron-sulfur clusters used by many enzymes. Disruptions in the assembly of Fe-S clusters is associated with Friedreich ataxia and Parkinson disease. Recent work has identified mutations in FdxR in patients with neuropathic hearing loss and visual impairment, somewhat resembling the global neurologic disorders seen with mitochondrial diseases. Impaired steroidogenesis is to be expected in such individuals, but this has not yet been studied.
Keyword
Figure
-
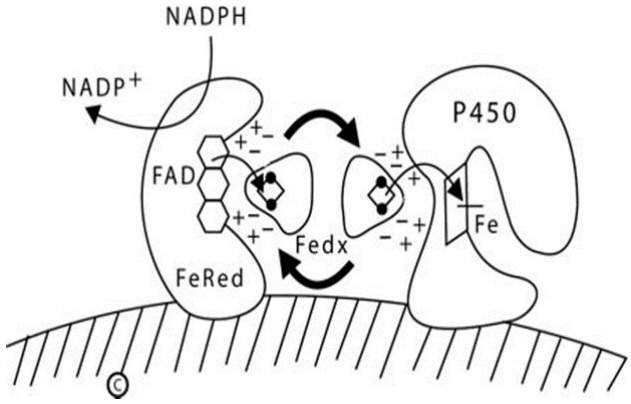
Fig. 1. Diagram of type 1 (mitochondrial) P450 enzymes. The inner mitochondrial membrane is indicated by the hatched area; both ferredoxin reductase (FeRed) and the P450 are membrane bound, but ferredoxin (Fedx) is not. NADPH donates a pair of electrons to the flavin adenine dinucleotide (FAD) moiety of ferredoxin reductase; which then donates them to the 2Fe2S center of ferredoxin (depicted by the ball-and-stick image). The same surface of the ferredoxin molecule interacts with both the FAD of ferredoxin reductase and the redox partner binding site of the P450 by electrostatic (charge-charge) interactions. Ferredoxin thus acts as an indiscriminate electron-shuttling protein that can support the catalysis of any available type 1 P450. The electrons reach the heme iron of the P450 permitting catalysis. NADP+, nicotinamide adenine dinucleotide phosphate NADPH, reduced adenine dinucleotide phosphate.

Fig. 2. Diagram of type 2 (microsomal) P450 enzymes. The endoplasmic reticulum membrane is indicated by the hatched area; both P450 oxidoreductase (POR) and the P450 are membrane-bound. NADPH interacts with the flavin adenine dinucleotide (FAD) domain of POR and donates a pair of electrons to the FAD moiety. Electron receipt elicits a conformational change, permitting the isoalloxazine rings of the FAD and flavin mononucleotide (FMN) moieties to come close together, permitting the electrons to transfer from the FAD to the FMN. Electron receipt by the FMN reverts the POR protein to its original, open conformation, permitting the FMN domain to interact with the redox partner binding site of the P450 by electrostatic charge interactions. The electrons reach the iron atom of the heme group of the P450, permitting catalysis. For some reactions catalyzed by some P450 enzymes, notably the 17,20-lyase activity of human P450c17, cytochrome b5 acts allosterically to promote increased activity. NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced adenine dinucleotide phosphate.
Reference
-
References
1. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011; 32:81–151.
Article2. Kim CJ. Congenital lipoid adrenal hyperplasia. Ann Pediatr Endocrinol Metab. 2014; 19:179–83.
Article3. Sahakitrungruang T. Clinical and molecular review of atypical congenital adrenal hyperplasia. Ann Pediatr Endocrinol Metab. 2015; 20:1–7.
Article4. Choi JH, Kim GH, Yoo HW. Recent advances in biochemical and molecular analysis of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Ann Pediatr Endocrinol Metab. 2016; 21:1–6.
Article5. Miller WL. Disorders in the initial steps in adrenal steroido - genesis. J Steroid Biochem Mol Biol. 2017; 165:18–37.6. Miller WL. Genetic disorders of vitamin D biosynthesis and degradation. J Steroid Biochem Mol Biol. 2017; 165:101–8.
Article7. Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev. 1988; 9:295–318.
Article8. Voutilainen R, Tapanainen J, Chung B, Matteson KJ, Miller WL. Hormonal regulation of P450scc (20,22 desmolase) and P450c17 (17α-hydroxylase/17,20 lyase) in cultured human granulosa cells. J Clin Endocrinol Metab. 1986; 63:202–7.9. Miller WL, Bose HS. Early steps in steroidogenesis: Intracellular cholesterol trafficking. J Lipid Res. 2011; 52:2111–35.
Article10. Bose HS, Lingappa VR, Miller WL. Rapid regulation of steroidogenesis by mitochondrial protein import. Nature. 2002; 417:87–91.
Article11. Toaff ME, Schleyer H, Strauss JF 3rd. Metabolism of 25-hydroxycholesterol by rat luteal mitochondria and dispersed cells. Endocrinology. 1982; 111:1785–90.
Article12. Bose HS, Whittal RM, Huang MC, Baldwin MA, Miller WL. N-218 MLN64, a protein with StAR-like steroidogenic ac t iv ity is folde d and cle ave d simi l arly to StAR. Biochemistry. 2000; 39:11722–31.
Article13. Cherradi N, Rossier MF, Vallotton MB, Timberg R, Friedberg I, Orly J, et al. Submitochondrial distribution of three key steroidogenic proteins (steroidogenic acute regulatory protein and cytochrome P450scc and 3β-hydroxysteroid dehydrogenase isomerase enzymes) upon stimulation by intracellular calcium in adrenal glomerulosa cells. J Biol Chem. 1997; 272:7899–907.
Article14. Ortiz de Monellano PR. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem Rev. 2010; 110:932–48.
Article15. Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. P450 Enzymes: their structure, reactivity, and selectivity—modeled by QM/MM Calculations. Chem Rev. 2010; 110:949–1017.
Article16. Nebert DW, Wikvall K, Miller WL. Human cytochromes P450 in health and disease. Phil Trans Roy S oc B. 2013; 368:20120431.
Article17. Miller WL. Minireview. Regulation of steroidogenesis by electron transfer. Endocrinology. 2005; 146:2544–50.
Article18. Solish SB, Picado-Leonard J, Morel Y, Kuhn RW, Mohandas TK, Hanukoglu I, et al. Human adrenodoxin reductase: two mRNAs encoded by a single gene on chromosome 17cen→q25 are expressed in steroidogenic tissues. Proc Natl Acad Sci USA. 1988; 85:7104–8.
Article19. Lin D, Shi Y, Miller WL. Cloning and sequence of the human adrenodoxin reductase gene. Proc Natl Acad Sci USA. 1990; 87:8516–20.
Article20. Coghlan VM, Vickery LE. Site-specific mutations in human ferredoxin that affect binding to ferredoxin reductase and cytochrome P450scc. J Biol Chem. 1991; 266:18606–12.
Article21. Brandt ME, Vickery LE. Expression and characterization of human mitochondrial ferredoxin reductase in Escherichia coli. Arch Biochem Biophys. 1992; 294:735–40.
Article22. Bak DW, Elliott SJ. Alternative FeS cluster ligands: tuning redox potentials and chemistry. Curr Opin Chem Biol. 2014; 19:50–8.
Article23. Braymer JJ, Lill R. Iron-sulfur biogenesis and trafficking in mitochondria. J Biol Chem. 2017; 292:12754–63.24. Maio N, Rouault TA. Outlining the complex pathway of mammalian Fe-S cluster biogenesis. Trends Biochem Sci. 2020; 45:411–26.
Article25. Maio N, Jain A, Rouault TA. Mammalian iron–sulfur cluster biogenesis: recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr Opin Chem Biol. 2020; 55:34–44.
Article26. Zhang W, Xu L, Zhao H, Li K. Mammalian mitochondrial iron–sulfur cluster biogenesis and transfer and related human diseases. Biophys Rep. 2021; 7:127–41.
Article27. Rouault TA. Mammalian iron–sulphur proteins: novel insights into biogenesis and function. Nature Rev Mol Cell Biol. 2015; 16:45–55.
Article28. Stehling O, Lill R. The role of mitochondria in cellular iron–sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harbor Perspect Biol. 2013; 5:a011312.
Article29. Isaya G. Mitochondrial iron–sulfur cluster dysfunction in neurodegenerative disease. Front Pharmacol. 2014; 5:29.
Article30. Valentine RC. Bacterial ferredoxin. Bacteriol Rev. 1964; 28:497–517.
Article31. Chashchin VL, Lapko VN, Adamovich TB, Kirillova NM, Lapko AG, Akhrem AA. The primary structure of hepatoredoxin from bovine liver mitochondria. Bioorg Khim. 1986; 12:1286–9.32. Okamura T, John ME, Zuber MX, Simpson ER, Waterman MR. Molecular cloning and amino acid sequence of the precursor form of bovine adrenodoxin. Evidence for a previously unidentified COOH-terminal peptide. Proc Natl Acad Sci USA. 1985; 82:5705–9.
Article33. Picado-Leonard J, Voutilainen R, Kao L, Chung B, Strauss JF 3rd, Miller WL. Human adrenodoxin: cloning of three cDNAs and cycloheximide enhancement in JEG-3 cells. J Biol Chem. 1988; 263:3240–4.
Article34. Mittal S, Zhu YZ, Vickery LE. Molecular cloning and sequence analysis of human placental ferredoxin. Arch Biochem Biophys. 1988; 264:383–91.
Article35. Chang CY, Wu DA, Lai CC, Miller WL, Chung BC. Cloning and structure of the human adrenodoxin gene. DNA. 1988; 7:609–15.
Article36. Morel Y, Picado-Leonard J, Wu DA, Chang C, Mohandas TK, Chung B, et al. Assignment of the functional gene for adrenodoxin to chromosome 11q13→qter and of two adrenodoxin pseudogenes to chromosome 20cen→q13.1. Am J Hum Genet. 1988; 43:52–9.37. Sparkes RS, Klisak I, Miller WL. Regional mapping of genes encoding human steroidogenic enzymes: P450scc to 15q23-q24, adrenodoxin to 11q22, adrenodoxin reductase to 17q24-q25, and P450c17 to 10q24-q25. DNA Cell Biol. 1991; 10:359–65.
Article38. Qi W, Li J, Cowan JA. Human ferredoxin-2 displays a unique conformational change. Dalton Trans. 2013; 42:3088–91.
Article39. Sheftel AD, Stehling O, Pierik AJ, Elsasser HP, Muhlenhoff U, Webert H, et al. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci USA. 2010; 107:11775–80.
Article40. Shi Y, Ghosh M, Kovtunovych G, Crooks DR, Rouault TA. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim Biophys Acta. 2012; 1823:484–92.
Article41. Cai K, Tonelli M, Frederick RO, Markley JL. Human mitochondrial ferredoxin 1 (FDX1) and ferredoxin 2 (FDX2) both bind cysteine desulfurase and donate electrons for iron-sulfur cluster biosynthesis. Biochemistry. 2017; 56:487–99.
Article42. Griffin A, Parajes S, Weger M, Zaucker A, Taylor AE, O’Neil DM, et al. Ferredoxin 1b (Fdx1b) is the essential mitochondrial redox partner for cortisol biosynthesis in zebrafish. Endocrinology. 2016; 157:1122–34.
Article43. Oakes JA, Li N, Wistow BRC, Griffin A, Barnard L, Storbeck KH, et al. Ferredoxin 1b deficiency leads to testis disorganization, impaired spermatogenesis and feminization in zebrafish. Endocrinology. 2019; 160:2401–16.
Article44. Spiegel R, Saada A, Halvardson J, Soiferman D, Shaag A, Edvardson S, et al. Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Euro J Hum Genet. 2014; 22:902–6.
Article45. Gurgel-Giannetti J, Lynch DS, Paiva ARB, Lucato LT, Yamamoto G, Thomsen C, et al. A novel complex neurological phenotype due to a homozygous mutation in FDX2. Brain. 2018; 141:2289–98.
Article46. Brentano ST, Black SM, Lin D, Miller WL. cAMP post-transcriptionally diminishes the abundance of adrenodoxin reductase mRNA. Proc Natl Acad Sci USA. 1992; 89:4099–103.
Article47. Paul A, Drecourt A, Petit F, Deguine DD, Vasnier C, Oufadem M, et al. FDXR mutations cause sensorial neuropathies and expand the spectrum of mitochondrial Fe-S-synthesis diseases. Am J Hum Genet. 2017; 101:630–7.
Article48. Peng Y, Shinde DN, Valencia CA, Mo JS, Rosenfeld J, Truitt Cho M, et al. Biallelic mutations in the ferredoxin reductase gene cause novel mitochondriopathy with optic atrophy. Hum Mol Genet. 2017; 26:4937–50.
Article49. Slone J, Peng Y, Chamberlin A, Harris B, Kaylor J, McDonald MT, et al. Biallelic mutations in FDXR cause neurodegeneration associated with inflammation. J Hum Genet. 2018; 63:1211–22.
Article50. Slone JD, Yang L, Yan Peng Y, Queme LF, Harris B, Rizzo SJS, et al. Integrated analysis of the molecular pathogenesis of FDXR-associated disease. Cell Death and Disease. 2020; 11:423.
Article51. Stenton SL, Piekutowska-Abramczuk D, Kulterer L, Kopajtich R, Claeys KG, Ciara E, et al. Expanding the clinical and genetic spectrum of FDXR deficiency by functional validation of variants of uncertain significance. Human Mutation. 2021; 42:310–9.
Article52. Jurkute N, Shanmugarajah PD, Hadjivassiliou M, Higgs J, Vojcic M, Horrocks I, et al. Expanding the FDXR-associated disease phenotype: retinal dystrophy is a recurrent ocular feature. Invest Ophthalmol Vis Sci. 2021; 62:2.
Article53. Fairfield H, Srivastava A, Ananda G, Liu R, Kircher M, Lakshminarayana A, et al. Exome sequencing reveals pathogenic mutations in 91 strains of mice with Mendelian disorders. Genome Res. 2015; 25:948–57.
Article54. Baker BY, Lin L, Kim CJ, Raza J, Smith CP, Miller WL, et al. Non-classic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab. 2006; 91:4781–5.
Article55. Sahakitrungruang T, Tee MK, Blackett PR, Miller WL. Partial defect in the cholesterol side-chain cleavage enzyme, P450scc (CYP11A1) resembling non-classic congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab. 2011; 96:792–8.56. Pandey AV, Flück CE. NADPH P450 oxidoreductase: structure, function, and pathology of diseases. Pharmacol Therap. 2013; 138:229–54.
Article57. Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci USA. 1997; 94:8411–6.58. Ellis J, Gutierrez A, Barsukov IL, Huang WC, Grossmann JG, Roberts GC. Domain motion in cytochrome P450 reductase: conformational equilibria revealed by NMR and small-angle x-ray scattering. J Biol Chem. 2009; 284:36628–37.59. Shen AL, O'Leary KA, Kasper CB. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2002; 277:6536–41.
Article60. Otto DM, Henderson CJ, Carrie D, Davey M, Gundersen TE, Blomhoff R, et al. Identification of novel roles of the cytochrome P450 system in early embryogenesis: effects on vasculogenesis and retinoic acid homeostasis. Mol Cell Biol. 2003; 23:6103–16.
Article61. Flück CE, Tajima T, Pandey AV, Arlt W, Okuhara K, Verge CF, et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nature Genetics. 2004; 36:228–30.
Article62. Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, et al. Diversity and function of mutations in P450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005; 76:729–49.
Article63. Laue K, Pogoda HM, Daniel PB, van Haeringen A, Alanay Y, von Ameln S, et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011; 89:595–606.
Article64. Hershkovitz E, Parvari R, Wudy SA, Hartmann MF, Gomes LG, Loewental N, et al. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20 lyase deficiency. J Clin Endocrinol Metab. 2008; 93:3584–8.65. Sahakitrungruang T, Huang N, Tee MK, Agrawal V, Russell WE, Crock P, et al. Clinical, genetic and enzymatic characterization of P450 oxidoreductase deficiency in four patients. J Clin Endocrinol Metab. 2009; 94:4992–5000.
Article66. Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, et al. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab. 2005; 90:414–26.
Article67. Krone N, Reisch N, Idkowiak J, Dhir V, Ivison HE, Hughes BA, et al. Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2012; 97:E257–67.68. Geller DH, Auchus RJ, Mendonça BB, Miller WL. The genetic and functional basis of isolated 17,20 lyase deficiency. Nature Genet. 1997; 17:201–5.69. Geller DH, Auchus RJ, Miller WL. P450c17 mutations R347H and R358Q selectively disrupt 17,20 lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5. Mol Endocrinol. 1999; 13:167–75.70. Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A. Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am J Med Genet A. 2004; 128A:333–9.71. Fukami M, Hasegawa T, Horikawa R, Ohashi T, Nishimura G, Homma K, et al. Cytochrome P450 oxidoreductase deficiency in three patients initially regarded as having 21-hydroxylase deficiency and/or aromatase deficiency: diagnostic value of urine steroid hormone analysis. Pediatr Res. 2006; 59:276–80.
Article72. Shackleton C, Marcos J, Arlt W, Hauffa BP. Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): a disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype. Am J Med Genet A. 2004; 129A:105–12.
Article73. Pandey AV, Kempna P, Hofer G, Mullis PE, Flück CE. 2007. Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Mol Endocrinol. 2007; 21:2579–95.74. Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004; 15:432–8.
Article75. Homma K, Hasegawa T, Nagai T, Adachi M, Horikawa R, Fujiwara I, et al. Urine steroid hormone profile analysis in cytochrome P450 oxidoreductase deficiency: implication for the backdoor pathway to dihydrotestosterone. J Clin Endocrinol Metab. 2006; 91:2643–9.
Article76. Huang N, Agrawal V, Giacomini KM, Miller WL. Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutants. Proc Natl Acad Sci USA. 2008; 105:1733–8.77. O’Leary KA, Li HC, Ram PA, McQuiddy P, Waxman DJ, Kasper CB. Thyroid regulation of NADPH:cytochrome P450 oxidoreductase: identification of a thyroid-responsive element in the 5’-flank of the oxidoreductase gene. Mol Pharmacol. 1997; 52:46–53.
Article78. Li HC, Liu D, Waxman DJ. Transcriptional induction of hepatic NADPH: cytochrome P450 oxidoreductase by thyroid hormone. Mol Pharmacol. 2001; 59:987–95.
Article79. Tee MK, Huang N, Damm I, Miller WL. Transcriptional regulation of human P450 oxidoreductase: identification of transcription factors and influence of promoter polymorphisms. Mol Endocrinol. 2011; 25:715–31.80. Gomes LG, Huang N, Agrawal V, Mendonca BB, Bachega TASS, Miller WL. The common P450 oxidoreductase variant A503V is not a modifier gene for 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2008; 93:2913–6.
Article81. Agrawal V, Huang N, Miller WL. Pharmacogenetics of P450 oxidoreductase. Effect of sequence variants on activities of CYP1A2 and CYP2C19. Pharmacogenet Genomics. 2008; 18:569–76.
Article82. Ingelman-Sundberg M. Polymorphism of cytochrome P450 and xenobiotic toxicity. Toxicology. 2002; 181-182:447–52.
Article83. Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003; 348:529–37.
Article84. Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, et al. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004; 305:683–6.
Article85. Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci USA. 2006; 103:13682–7.
Article86. Agrawal V, Choi JH, Giacomini KM, Miller WL. Substrate-specific modulation of CYP3A4 activity by genetic variants of cytochrome P450 oxidoreductase (POR). Pharmacogenet Genomics. 2010; 20:611–8.87. Sandee D, Morrissey K, Agrawal V, Tam HK, Kramer MA, Tracy TS, et al. Effects of genetic variants of P450 oxidoreductase on catalysis by CYP2D6 in vitro. Pharmacogenet Genomics. 2010; 20:677–86.88. Miller WL, Agrawal V, Sandee D, Tee MK, Huang N, Choi JH, et al. Consequences of POR mutations and polymorphisms. Mol Cell Endocrinol. 2011; 336:174–9.
Article89. Giordano S, Steggles A. The human liver and reticulocyte cytochrome b5 mRNA's are products from a single gene. Biochim Biophys Res Commun. 1991; 178:38–44.
Article90. Shephard EA, Povey S, Spurr NK, Phillips IR. Chromosomal localization of a cytochrome b5 gene to human chromosome 18 and a cytochrome b5 pseudogene to the X chromosome. Genomics. 1992; 11:302–8.
Article91. Storbeck KH, Swart AC, Fox CL, Swart P. Cytochrome b5 modulates multiple reactions in steroidogenesis by diverse mechanisms. J Steroid Biochem Mol Biol. 2015; 151:66–73.
Article92. Bridges A, Gruenke L, Chang YT, Vakser IA, Loew GH, Waskell L. Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem. 1998; 273:17036–49.93. Yamazaki H, Johnson WW, Ueng YF, Shimada T, Guengerich FP. Lack of electron transfer from cytochrome b5 in stimulation of catalytic activities of cytochrome P450 3A4. Characterization of a reconstituted cytochrome P450 3A4/NADPH-cytochrome P450 reductase system and studies of apo-cytochrome b5. J Biol Chem. 1996; 271:27438–44.94. Auchus RJ, Lee TC, Miller WL. Cytochrome b5 augments the 17,20 lyase activity of human P450c17 without direct electron transfer. J Biol Chem. 1998; 273:3158–65.95. Pandey AV, Miller WL. Regulation of 17,20 lyase activity by cytochrome b5 and by serine phosphorylation of P450c17. J Biol Chem. 2005; 280:13265–71.
Article96. Yanagibashi K, Hall P. Role of electron transport in the regulation of the lyase activity of C21 side-chain cleavage P-450 from porcine adrenal and testicular microsomes. J Biol Chem. 1986; 26:8429–33.
Article97. Lin D, Black SM, Nagahama Y, Miller WL. Steroid 17α-hydroxylase and 17,20 lyase activities of P450c17: contributions of serine106 and of P450 reductase. Endocrinology. 1993; 132:2498–506.
Article98. Lee - Robichaud P, Akhtar ME, Akhtar M. Lysine mutagenesis identifies cationic charges of human CYP17 that interact with cytochrome b5 to promote male sexhormone biosynthesis. Biochem J. 1999; 342:309–12.
Article99. Swart AC, Storbeck KH, Swart P. A single amino acid residue Ala 105, confers 16α-hydroxylase activity to human cytochrome P450 17α-hydroxylase/17,20 lyase. J Steroid Biochem Mol Biol. 2010; 119:112–20.
Article100. Sherbet DP, Tiosano D, Kwist KM, Hochberg Z, Auchus RJ. CYP17 mutation E305G causes isolated 17,20 lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003; 278:48563–9.101. Zhang L, Rodriguez H, Ohno S, Miller WL. Serine phosphorylation of human P450c17 increases 17,20 lyase activity: implications for adrenarche and for the polycystic ovary syndrome. Proc Natl Acad Sci USA. 1995; 92:10619–23.102. Pandey AV, Mellon SH, Miller WL. Protein phosphatase 2A and phosphoprotein SET regulate androgen production by P450c17. J Biol Chem. 2003; 278:2837–44.
Article103. Tee MK, Miller WL. Phosphorylation of human cytochrome P450c17 by p38α selectively increases 17,20 lyase activity and androgen synthesis. J Biol Chem. 2013; 288:23903–13.104. Naffin-Olivos JL, Auchus RJ. Human c ytochrome b5 requires residues E48 and E49 to stimulate the 17,20-lyase activity of cytochrome P450c17. Biochemistry. 2006; 24:755–62.105. Miller WL. The syndrome of 17,20 lyase deficiency. J Clin Endocrinol Metab. 2012; 97:59–67.
Article106. Hegesh E, Hegesh J, Kaftory A. Congenital methemoglobinemia with deficiency of cytochrome b5. New England J Med. 1986; 314:757–61.107. Giordano SJ, Kaftory A, Steggles AW. A splicing mutation in the cytochrome b5 gene from a patient with congenital methemoglobinemia and pseudohermaphroditism. Human Genet. 1994; 93:568–70.108. Kok RC, Timmerman MA, Wolfenbuttel KP, Drop SLS, deJong FH. Isolated 17,20 lyase deficiency due to the cytochrome b5 mutation W27X. J Clin Endocrinol Metab. 2010; 95:994–9.109. Idkowiak J, Randell T, Dhir V, Patel P, Shackleton CH, Taylor NF, et al. A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J Clin Endocrinol Metab. 2012; 97:E465–75.
Article110. Leung MT, Cheung HN, Iu YP, Choi CH, Tiu SC, Shek CC. Isolated 17, 20-lyase deficiency in a CYB5A mutated female with normal sexual development and fertility. J Endocr Soc. 2020; 4:1–8.111. Shaunak M, Taylor NF, Hunt D, Davies JH. Isolated 17,20 lyase deficiency secondary to a novel CYB5A variant: comparison of steroid metabolomic findings with published cases provides diagnostic guidelines and greater insight into its biological role. Horm Res Paediatr. 2020; 93:483–95.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A study on dose distribution of small irradiation field in the electron therapy
- Factors associated with discharge of children from the emergency department after interfacility transfer
- The Effects of Keratinocyte-derived Factors Involved in Transforming the Morphologies of Melanocyte and in Translocating Melanosomes to Keratinocyte
- Factors Influencing Confidence in Performance Competence of Core Basic Nursing Skills by Nursing Students
- Recent advances in electron microscopy for the diagnosis and research of glomerular diseases
