Congenital hyperinsulinism: 2 case reports with different rare variants in ABCC8
- Mouron-Hryciuk J
 1
1 - Stoppa-Vaucher S1,2
- Busiah K1
- Bouthors T1
- Antoniou MC1
- Jacot E3
- Brusgaard K4
- Christesen HT5
- Hussain K6
- Dwyer A7
- Roth-Kleiner M8
- Hauschild M1
- Affiliations
-
- 1Pediatric Endocrinology and Diabetology Unit, Ser vice of Pediatrics, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 2Department of Pediatrics, Hôpitaux Neuchâtelois, Neuchâtel, Switzerland
- 3Diabetology, Neuchâtel, Switzerland
- 4Departement of Clinical Genetics, Odense University Hospital, Odense, Denmark
- 5Hans Christian Andersen Children’s Hospital, Odense University Hospital, Odense, Denmark
- 6D e velopmental Endocr inology Research Group, Clinical and Molecular Genetics Unit, Institute of Child Health, University College London, London, UK
- 7Boston College, William F. Connell School of Nursing, Chestnut Hill, MA, USA
- 8Service of Neonatology, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- KMID: 2514154
- DOI: http://doi.org/10.6065/apem.2040042.021
Abstract
- Congenital hyperinsulinism (CHI) is a rare glucose metabolism disorder characterized by unregulated secretion of insulin that leads to hyperinsulinemic hypoglycemia (HH). Most cases are caused by mutations in the KATP-channel genes ABCC8 and KCNJ11. We report 2 patients that experienced severe HH from the first day of life. Patient 1 developed midgut volvulus after initiating diazoxide and required intestinal resection. He was subsequently managed with a high-dose octreotide and glucose-enriched diet. Consistent with diffuse type CHI by 18F-dihydroxyphenylalanine positron emission tomography-computed tomography, genetic testing revealed a homozygous ABCC8 variant, c.1801G>A, p.(Val601Ile). The rare variant was previously reported to be diazoxide-responsive, and the patient responded well to diazoxide monotherapy, with clinical remission at 2 years of age. Patient 2 responded to diazoxide with spontaneous clinical remission at 15 months of age. However, an oral glucose tolerance test at 7 years of age revealed hyperinsulinism. Genetic testing revealed that the proband and several seemingly healthy family members harbored a novel, heterozygous ABCC8 variant, c.1780T>C, p.(Ser594Pro). Genetic findings identified previously unrecognized HH in the proband’s mother. The proband’s uncle had been diagnosed with monogenic ABCC8-diabetes and was successfully transitioned from insulin to glibenclamide therapy. We report findings of intestinal malrotation and volvulus occurring 2 days after initiation of diazoxide treatment. We also report a novel, heterozygous ABCC8 variant in a family that exhibited cases of CHI in infancy and HH and monogenic diabetes in adult members. The cases demonstrate the importance and clinical utility of genetic analyses for informing and guiding treatment and care.
Figure
-
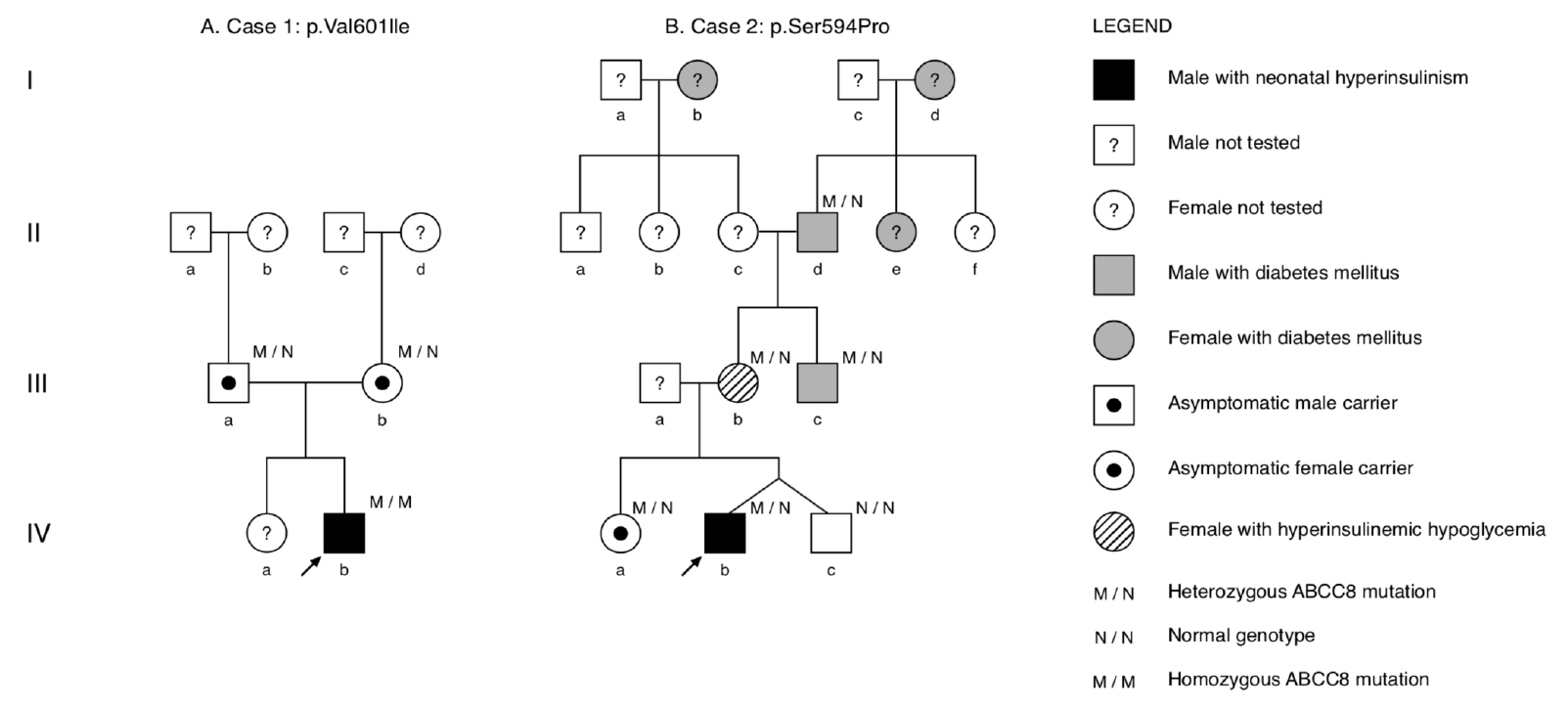
Fig. 1. Pedigrees showing inheritance of ABCC8 mutations in both families. Squares represent males and circles represent females. The arrow depicts the proband with CHI. The individual lVa of family B underwent genetic testing and was confirmed not to be a carrier of ABCC8 mutation. (A) Family A: parents (IIIa/b) are asymptomatic carriers. (B) Family B: variable presentation of individuals harboring the heterozygous ABCC8 mutation, including symptomatic hyperinsulinemic hypoglycemia (IIIb), monogenic diabetes (IIIc), and diabetes of unknown origin (IId).

Fig. 2. Axial fluorine-18L-3,4hydroxyphenylalanine positron emission tomography image. Diffuse uptake of F-fluoro-L-DOPA by the pancreas is visualized by the hot spot (white arrow). Physiological distribution of the radiotracer is observed with higher accumulation in the kidneys and lower accumulation in the liver.

Fig. 3. Three-dimensional modelled structure of a pancreatic ATP-sensitive potassium channel. (A) The KATP-channel is a hetero-octameric complex composed of 4 Kir6.2 subunits and 4 SUR1 units. The transmembrane domains are highlighted. (B) The genetic variants in our 2 cases are located on the same transmembrane domain (TMD1) of the SUR1 subunit.
Reference
-
References
1. Bellanné-Chantelot C, Saint-Martin C, Ribeiro MJ, Vaury C, Verkarre V, Arnoux JB, et al. ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism. J Med Genet. 2010; 47:752–9.
Article2. Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013; 168:557–64.
Article3. Roženková K, Güemes M, Shah P, Hussain K. The diagnosis and management of hyperinsulinaemic hypoglycaemia. J Clin Res Pediatr Endocrinol. 2015; 7:86–97.
Article4. Helleskov A, Melikyan M, Globa E, Shcherderkina I, Poertner F, Larsen AM, et al. Both low blood glucose and insufficient treatment confer risk of neurodevelopmental impairment in congenital hyperinsulinism: a multinational cohort study. Front Endocrinol. 2017; 8:156.
Article5. R asmussen AG, Melikian M, Globa E, Detlefsen S, Rasmussen L, Petersen H, et al. The difficult management of persistent, non-focal congenital hyperinsulinism: a retrospective review from a single, tertiary center. Pediatr Diabetes. 2020; 21:441–55.
Article6. Yorifuji T. Congenital hyperinsulinism: current status and future perspectives. Ann Pediatr Endocrinol Metab. 2014; 19:57–68.
Article7. Gillis D. Familial hyperinsulinism. In : Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle;1993-2020. [cited 2019 Nov 20]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1375/.8. Vajravelu ME, De León DD. Genetic characteristics of patients with congenital hyperinsulinism. Curr Opin Pediatr. 2018; 30:568–75.
Article9. Flanagan SE, Kapoor RR, Hussain K. Genetics of congenital hyperinsulinemic hypoglycemia. Semin Pediatr Surg. 2011; 20:13–7.
Article10. Martínez R, Fernández-Ramos C, Vela A, Velayos T, Aguayo A, Urrutia I, et al. Clinical and genetic characterization of congenital hyperinsulinism in Spain. Eur J Endocrinol. 2016; 174:717–26.
Article11. Mohamed Z, Arya VB, Hussain K. Hyperinsulinaemic hypoglycaemia:genetic mechanisms, diagnosis and management. J Clin Res Pediatr Endocrinol. 2012; 4:169–8.12. Rahman SA, Nessa A, Hussain K. Molecular mechanisms of congenital hyperinsulinism. J Mol Endocrinol. 2015; 54:R119–29.
Article13. Misra S, Owen KR. Genetics of monogenic diabetes: present clinical challenges. Curr Diab Rep. 2018; 18:141.
Article14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. 2015; 17:405–24.
Article15. Arya VB, Guemes M, Nessa A, Alam S, Shah P, Gilbert C, et al. Clinical and histological heterogeneity of congenital hyperinsulinism due to paternally inherited heterozygous ABCC8/KCNJ11 mutations. Eur J Endocrinol. 2014; 171:685–95.
Article16. Strouse PJ. Disorders of intestinal rotation and fixation (« malrotation »). Pediatr Radiol. 2004; 34:837–51.
Article17. Salehi Karlslätt K, Pettersson M, Jäntti N, Szafranski P, Wester T, Husberg B, et al. Rare copy number variants contribute pathogenic alleles in patients with intestinal malrotation. Mol Genet Genomic Med. 2019; 7:e549.
Article18. Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011; 54:2575–83.
Article19. Vieira TC, Bergamin CS, Gurgel LC, Moisés RS. Hyperinsulinemic hypoglycemia evolving to gestational diabetes and diabetes mellitus in a family carrying the inactivating ABCC8 E1506K mutation. Pediatr Diabetes. 2010; 11:505–8.
Article20. Ortiz D, Bryan J. Neonatal diabetes and congenital hyperinsulinism caused by mutations in ABCC8/SUR1 are associated with altered and opposite affinities for ATP and ADP. Front Endocrinol. 2015; 6:48.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A novel mutation of ABCC8 gene in a patient with diazoxide-unresponsive congenital hyperinsulinism
- Pasireotide treatment for severe congenital hyperinsulinism due to a homozygous ABCC8 mutation
- Using low-dose octreotide with diazoxide-resistant congenital hyperinsulinism resulting from compound heterozygous mutations in the ABCC8 gene
- Congenital hyperinsulinism: current status and future perspectives
- A Case of Hyperinsulinism/hyperammonemia Syndrome
