Muscle pathology in neuromuscular disorders
- Affiliations
-
- 1Department of Neurology, Pusan National University School of Medicine, Yangsan, Korea
- 2Biomedical Research Institute, Pusan National University Hospital, Busan, Korea
- 3Biomedical Research Institute, Pusan National University Yangsan Hospital, Yangsan, Korea
- KMID: 2511079
- DOI: http://doi.org/10.14253/acn.2020.22.2.51
Abstract
- Muscle pathology findings may guide the diagnosis of neuromuscular disorders since they are helpful for understanding the pathological processes causing muscle weakness and also provide significant clues for the diagnosis of muscle diseases. Recent advances in molecular genetics mean that a muscle biopsy can be omitted when diagnosing inherited muscle diseases. However, the muscle pathology can still play a role in those cases and its findings are also required when diagnosing inflammatory myopathies.
Keyword
Figure
-
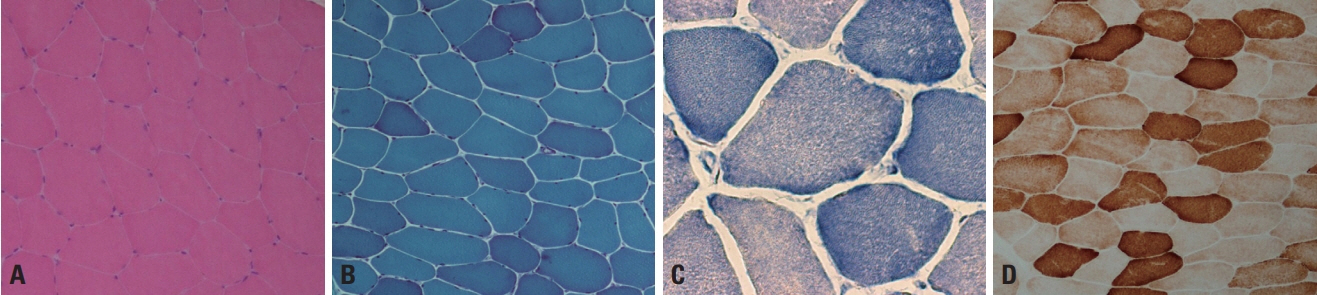
Fig. 1. Normal muscle pathology. (A, B) Hematoxylin and eosin (H&E) and modified Gomori trichrome (mGT) stains reveal polygonal shaped, multinucleated muscle fibers with minimal amounts of endomysial connective tissue (×200, respectively). (C) Nicotinamide dehydrogenase-tetrazolium reductase (NADH-TR) stain reveals the network arrangement of the myofibrils in a high-power field (×400). (D) Cytochrome c oxidase (COX) staining shows that the sarcoplasm is homogeneously stained in normal muscle fibers (×200).
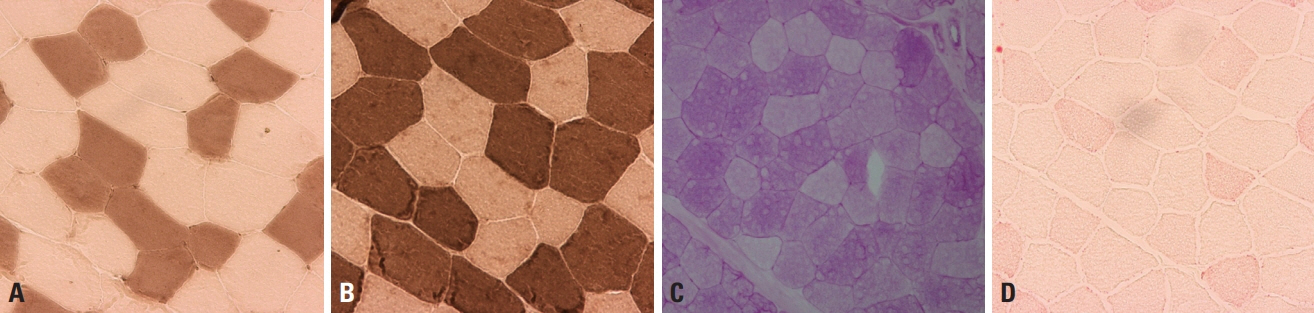
Fig. 2. Normal muscle pathology. (A, B) Adenosine triphosphatase (ATPase) stain differentiates type-1 and -2 fibers, with type-1 fibers being darkly stained at pH 10.6 (A, ×200) and lightly stained at pH 4.3 (B, ×200). (C) Periodic acid–Schiff (PAS) stain reflects the glycogen content, and is more prominent in type-2 fibers (×200). (D) Oil red O (ORO) stains lipid droplets within muscle fibers, and type-1 fibers are more evident since their lipid content is denser than in type-2 fibers (×200).
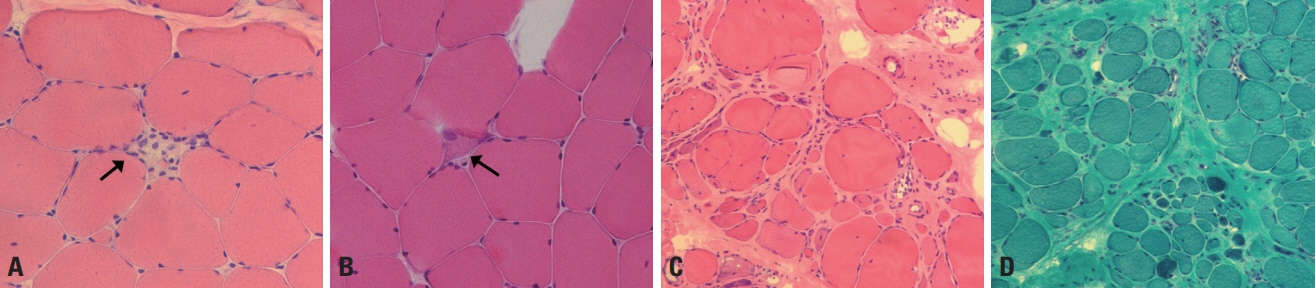
Fig. 3. Muscle pathology of muscular dystrophy. (A) A necrotic fiber (arrow) has a pale-colored cytoplasm and is invaded by macrophages (hematoxylin and eosin [H&E], ×200). (B) A regenerating fiber (arrow) has a basophilic cytoplasm with enlarged nuclei (H&E, ×200). (C) Atrophic and hypertrophic fibers are mixed, and the fibers are mostly round rather than polygonal in shape (H&E, ×100). (D) An increased amount of connective tissue fills up the spaces between muscle fibers (modified Gomori trichrome, ×100).
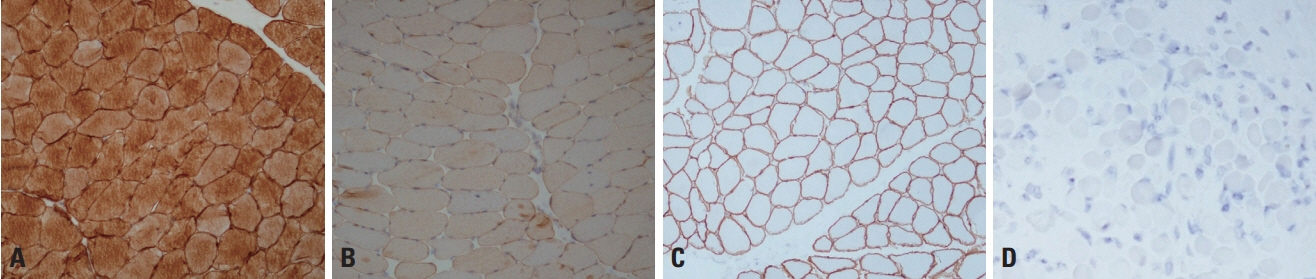
Fig. 4. Immunohistochemistry of dysferlinopathy and dystrophinopathy. (A, B) The antidysferlin antibody detects dysferlin in normal muscle pathology (A, ×100), but the staining is markedly decreased in dysferlinopathy (limb-girdle muscular dystrophy type 2B) (B, ×100). The antimerosin antibody demonstrates the localization of merosin to the sarcolemma (C, ×100), but staining is absent in merosin-deficient congenital muscular dystrophy type 1A (D, ×100).
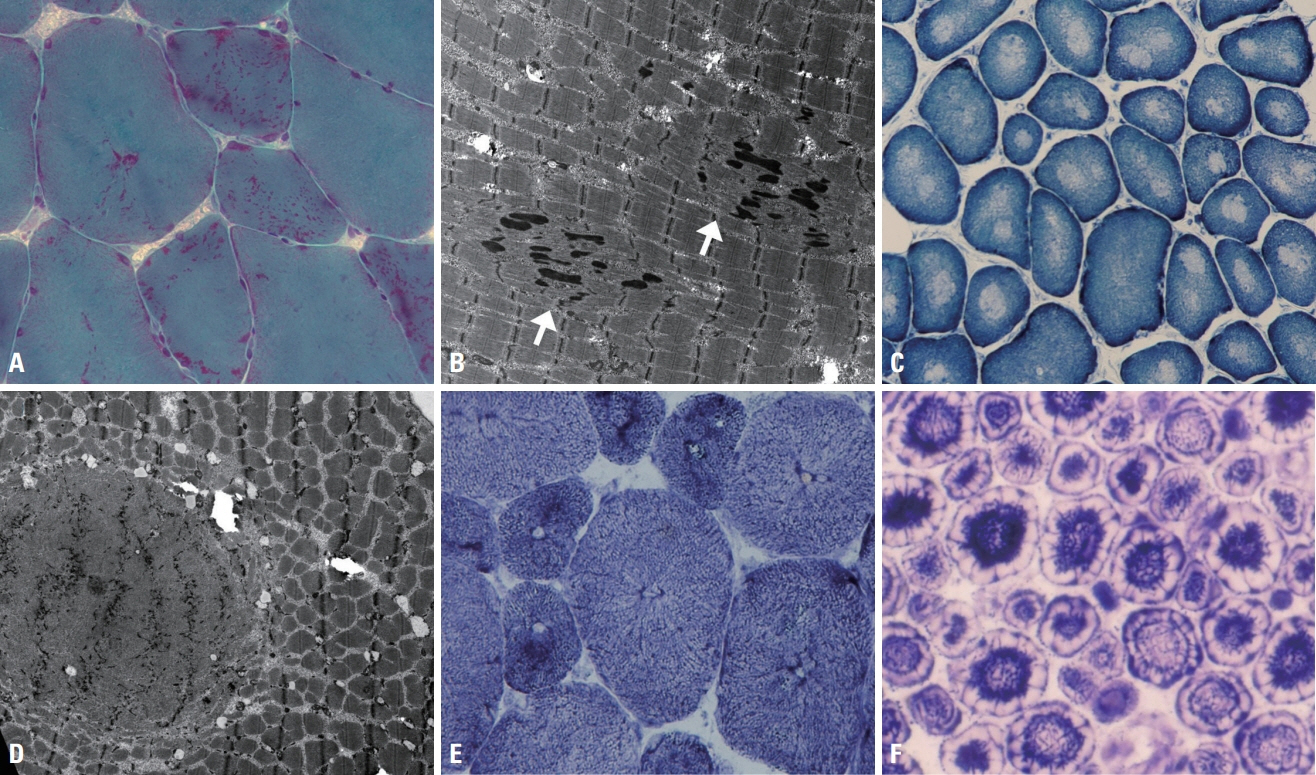
Fig. 5. Muscle pathology of congenital myopathies. (A) Nemaline bodies in nemaline myopathy appear as red with a dot-like shape, and are distributed in the sarcoplasm, the subsarcolemma, or both (modified Gomori trichrome, ×200). (B) Ultrastructurally the nemaline bodies appear rod-shaped with a similar density to Z-disks, and are associated with disorganized myofibrils (arrows) (×5,000). (C) In central core disease, nicotinamide dehydrogenase-tetrazolium reductase (NADH-TR) stain highlights the central cores with loss of the stain due to deficiency of the enzyme (×200). (D) The ultrastructural finding for the central core is a zone with disorganized myofibrillar structures (×4,000). (E) In centronuclear myopathy, radial arrangements of sarcoplasmic strands around central nuclei are revealed by NADH-TR stain (×400). (F) In myotubular myopathy, the peripheral regions of muscle fibers are deficient in oxidative enzyme due to immature myofibril formation, which produces a peripheral halo (×400).
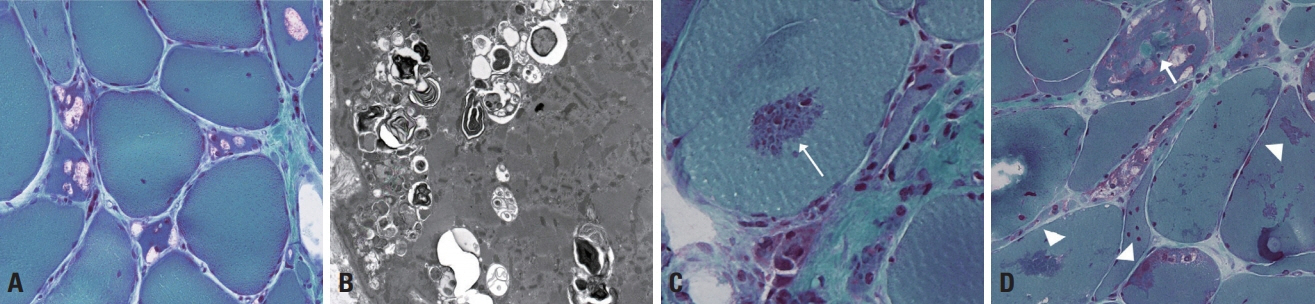
Fig. 6. Muscle pathology of GNE myopathy and myofibrillar myopathy. (A) Single or multiple rimmed vacuoles are present within muscle fibers in GNE myopathy (modified Gomori trichrome [mGT], ×200). (B) The ultrastructural finding is rimmed vacuoles appearing in clusters of autophagic vacuoles containing amorphous and myeloid structures (×6,000). (C) Red cytoplasmic bodies are present in myofibrillar myopathy (arrow) (mGT, ×400). (D) A spheroid body (arrow) appears round and green, and sarcoplasmic bodies (arrowheads) have irregular shapes and are green (mGT, ×200).
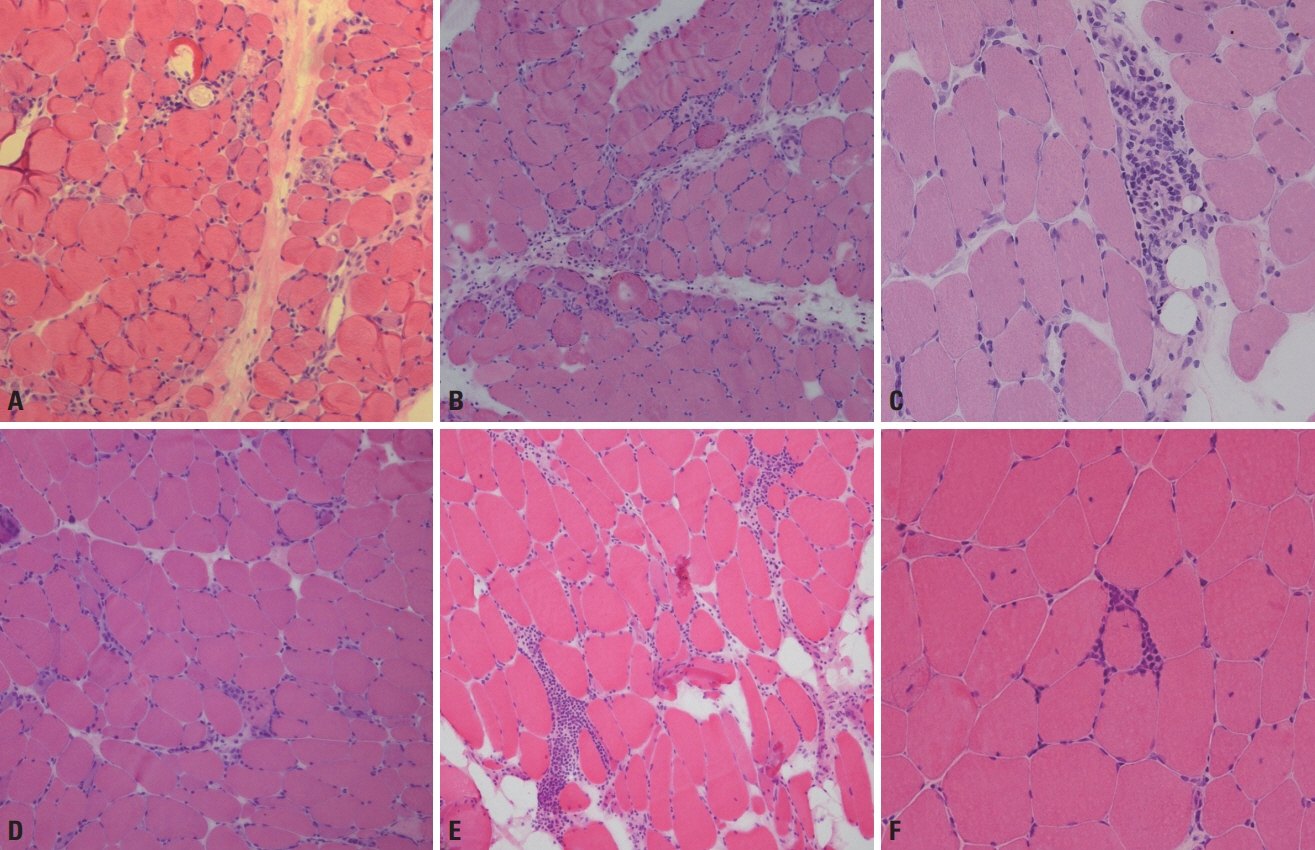
Fig. 7. Muscle pathology of idiopathic inflammatory myopathies. (A) Muscle fibers in perifascicular regions are more atrophied in dermatomyositis, which is called perifascicular atrophy (hematoxylin and eosin [H&E], ×100). (B) Necrotic fibers are frequent in perifascicular regions in antisynthetase syndrome, which is called perifasicular necrosis (H&E, ×100). (C) In dermatomyositis, inflammatory cellular infiltration is found in the perimysium and perivascular regions (H&E, ×200). (D) In immune-mediated necrotizing myopathy, necrotic and regenerating fibers appear frequently, whereas inflammatory infiltration rarely occurs (H&E, ×100). (E) Endomysial inflammatory infiltration is a characteristic of polymyositis (H&E, ×100). (F) In polymyositis, inflammatory cells surrounding or invading nonnecrotic fibers are frequently found (H&E, ×200).
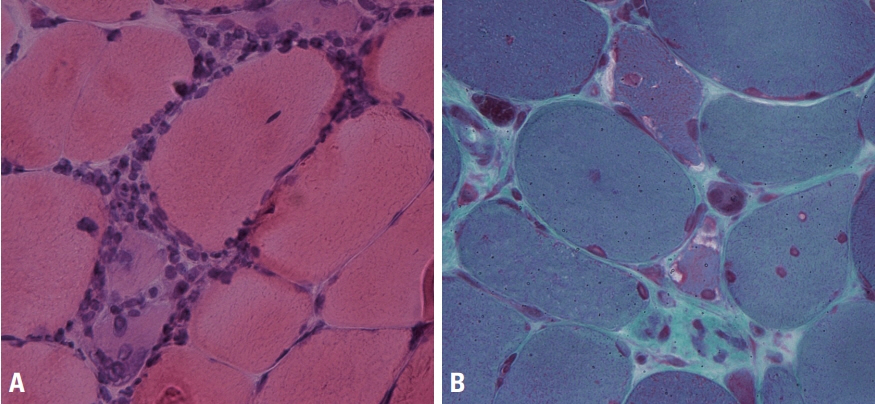
Fig. 8. Muscle pathology of inclusion-body myositis. (A) Inflammatory cells surround a necrotic muscle fiber and a normal-looking muscle fiber in inclusion-body myositis (hematoxylin and eosin, ×400). (B) Rimmed vacuoles are a characteristic finding of inclusion-body myositis (modified Gomori trichrome, ×400).
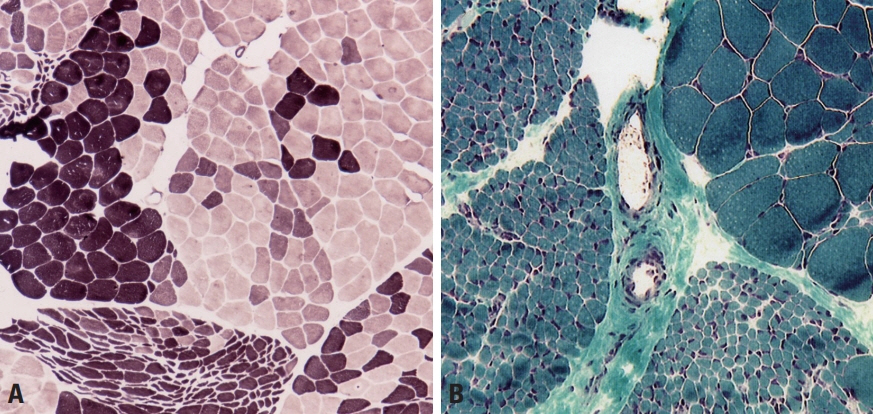
Fig. 9. Muscle pathology of neurogenic disorders. (A) In amyotrophic lateral sclerosis, darkly stained type-1 fibers and lightly stained type 2 fibers are grouped, rather than appearing mixed with a checkboard pattern (adenosine triphosphatase; pH 4.3, ×100). (B) Muscle fibers in a whole fascicle are atrophied in amyotrophic lateral sclerosis, which is called group atrophy or fascicular atrophy (modified Gomori trichrome, ×100).
Reference
-
1. Suzuki S, Uruha A, Suzuki N, Nishino I. Integrated diagnosis project for inflammatory myopathies: an association between autoantibodies and muscle pathology. Autoimmun Rev. 2017; 16:693–700.
Article2. Dubowitz V, Sewry CA, Oldfors A. Normal muscle. In : Dubowitz V, Sewry CA, Oldfors A, Lane R, editors. Muscle biopsy: a practical approach. 4th ed. London: Elsevier;2013. p. 28–54.3. Sewry CA, Goebel HH. General pathology of muscle disease. In : Goebel HH, Sewry CA, Weller RO, editors. Muscle disease: pathology and genetics. 2nd ed. Oxford: Wiley-Blackwell;2013. p. 19–38.4. Dubowitz V, Sewry CA, Oldfors A. Muscular dystrophies and allied disorders I: Duchenne and Becker muscular dystrophy. In : Dubowitz V, Sewry CA, Oldfors A, Lane R, editors. Muscle biopsy: a practical approach. 4th ed. London: Elsevier;2013. p. 250–275.5. Barresi R, Brown SC. Dystrophin and its associated glycoprotein complex. In : Goebel HH, Sewry CA, Weller RO, editors. Muscle disease: pathology and genetics. 2nd ed. Oxford: Wiley-Blackwell;2013. p. 95–101.6. Dubowitz V, Sewry CA. Muscular dystrophies and allied disorders IV: Emery-Dreifuss muscular dystrophy and similar syndromes. In : Dubowitz V, Sewry CA, Oldfors A, Lane R, editors. Muscle biopsy: a practical approach. 4th ed. London: Elsevier;2013. p. 331–344.7. Kim DS. Diagnostic significance of immunohistochemical staining in muscular dystrophy. J Korean Neurol Assoc. 2006; 24:1–13.8. Park YE, Shin JH, Kim HS, Lee CH, Kim DS. Characterization of congenital myopathies at a Korean neuromuscular center. Muscle Nerve. 2018; 58:235–244.
Article9. Ryan MM, Schnell C, Strickland CD, Shield LK, Morgan G, Iannaccone ST, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. 2001; 50:312–320.
Article10. Lee JM, Lim JG, Shin JH, Park YE, Kim DS. Clinical and genetic diversity of nemaline myopathy from a single neuromuscular center in Korea. J Neurol Sci. 2017; 383:61–68.
Article11. Wattanasirichaigoon D, Swoboda KJ, Takada F, Tong HQ, Lip V, Iannaccone ST, et al. Mutations of the slow muscle alpha-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology. 2002; 59:613–617.12. Park YE, Shin JH, Kang B, Lee CH, Kim DS. NEB-related core-rod myopathy with distinct clinical and pathological features. Muscle Nerve. 2016; 53:479–484.
Article13. Naddaf E, Milone M, Kansagra A, Buadi F, Kourelis T. Sporadic late-onset nemaline myopathy: clinical spectrum, survival, and treatment outcomes. Neurology. 2019; 93:e298–e305.14. Park YE, Choi YC, Bae JS, Lee CH, Kim HS, Shin JH, et al. Clinical and pathological features of Korean patients with DNM2-related centronuclear myopathy. J Clin Neurol. 2014; 10:24–31.15. Romero NB, Laforte J. Centronuclear myopathies. In : Goebel HH, Sewry CA, Weller RO, editors. Muscle disease: pathology and genetics. 2nd ed. Oxford: Wiley-Blackwell;2013. p. 134–144.16. Dubowitz V, Sewry CA. Myopathies with vacuoles. In : Dubowitz V, Sewry CA, Oldfors A, Lane R, editors. Muscle biopsy: a practical approach. 4th ed. London: Elsevier;2013. p. 406–422.17. Mair D, Biskup S, Kress W, Abicht A, Brück W, Zechel S, et al. Differential diagnosis of vacuolar myopathies in the NGS era. Brain Pathol. 2020; 30:877–896.
Article18. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975; 292:344–347.19. Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995; 38:705–713.
Article20. Tanboon J, Nishino I. Classification of idiopathic inflammatory myopathies: pathology perspectives. Curr Opin Neurol. 2019; 32:704–714.
Article21. Uruha A, Suzuki S, Suzuki N, Nishino I. Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain. 2016; 139(Pt 9):e50.
Article
