J Korean Med Sci.
2020 Aug;35(30):e252. 10.3346/jkms.2020.35.e252.
The First Case of an Infant with Familial A20 Haploinsufficiency in Korea
- Affiliations
-
- 1Department of Pediatrics, Medical Research Institute, Pusan National University School of Medicine, Pusan National University Hospital, Busan, Korea
- 2Department of Pediatrics, Pusan National University Children's Hospital, Yangsan, Korea
- 3Department of Dermatology, Pusan National University Yangsan Hospital, Yangsan, Korea
- 4Laboratory Medicine, Green Cross Genome, Yongin, Korea
- 5Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, Korea
- KMID: 2505192
- DOI: http://doi.org/10.3346/jkms.2020.35.e252
Abstract
- Haploinsufficiency of A20 (HA20) is a newly described autoinflammatory disease caused by loss-of-function mutations in the TNFAIP3 gene. Clinical phenotypes are heterogenous and resemble Behçet's disease, juvenile idiopathic arthritis, inflammatory bowel disease, or periodic fever syndrome, with symptoms developing at an early age. Here, we report the first case of infantile familial HA20 in Korea, which mimics neonatal lupus erythematosus (NLE). A 2-month-old infant exhibited symptoms including recurrent fever, erythematous rashes, and oral ulcers, with elevated liver enzymes, and tested positive for several autoantibodies, similar to systemic lupus erythematosus (SLE); therefore, she was suspected to have NLE. However, six months after birth, symptoms and autoantibodies persisted. Then, we considered the possibility of other diseases that could cause early onset rashes and abnormal autoantibodies, including autoinflammatory syndrome, monogenic SLE, or complement deficiency, all of which are rare. The detailed family history revealed that her father had recurrent symptoms, including oral and genital ulcers, knee arthralgia, abdominal pain, and diarrhea. These Behcet-like symptoms last for many years since he was a teenager, and he takes medications irregularly only when those are severe, but doesn't want the full-scale treatment. Whole-exome sequencing was conducted to identify a possible genetic disorder, which manifested as pathogenic variant nonsense mutation in the TNFAIP3 gene, leading to HA20. In conclusion, HA20 should be considered in the differential diagnosis of an infant with an early-onset dominantly inherited inflammatory disease that presents with recurrent oral and genital ulcerations and fluctuating autoantibodies. Additionally, it also should be considered in an infant with suspected NLE, whose symptoms and abnormal autoantibodies persist.
Figure
-
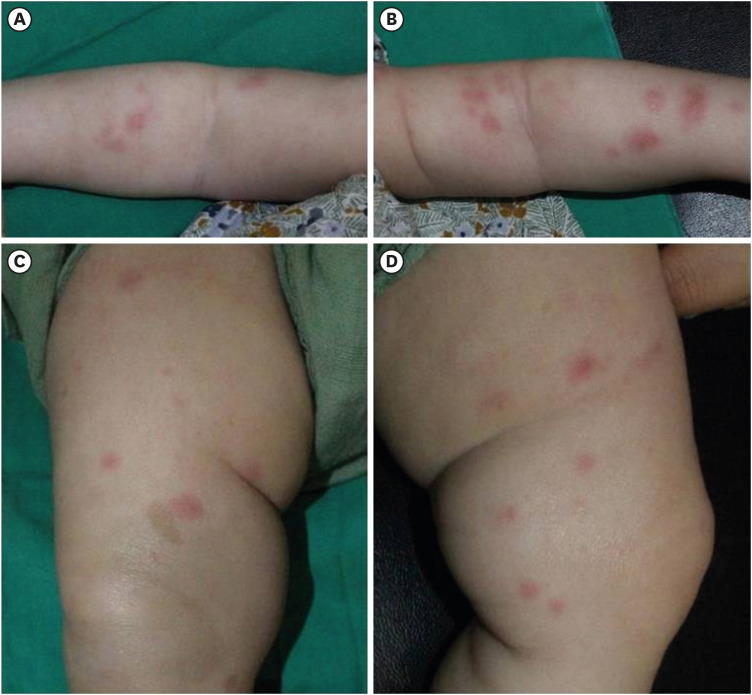
Fig. 1 Figures show erythematous wheal-like patches on the whole body and 4 extremities (A-D). You can also see the café au lait spot on (C). Figures are published under Informed consent.
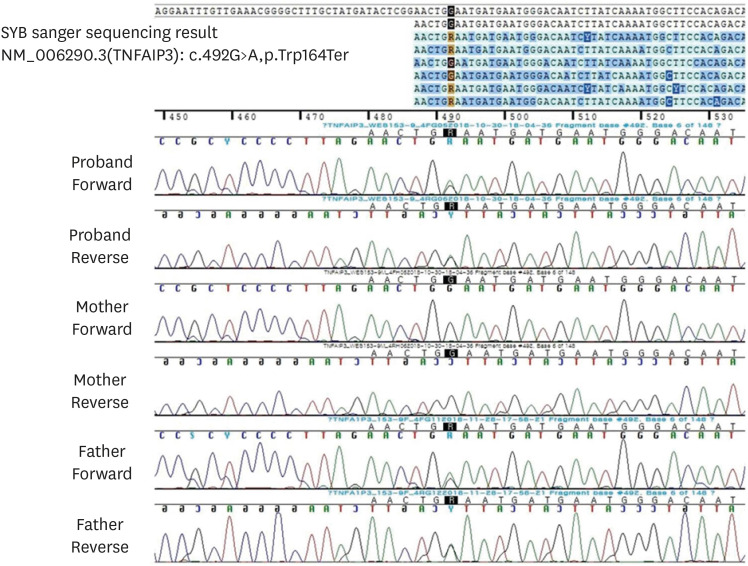
Fig. 2 Sanger sequencing analysis of the candidate variant in the TNFAIP3 gene of the family. The c.492G>A variation of the proband was inherited from her father.
Reference
-
1. Johnson AF, Nguyen HT, Veitia RA. Causes and effects of haploinsufficiency. Biol Rev Camb Philos Soc. 2019; 94(5):1774–1785. PMID: 31149781.
Article2. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2016; 48(1):67–73. PMID: 26642243.3. Aeschlimann FA, Laxer RM. Haploinsufficiency of A20 and other paediatric inflammatory disorders with mucosal involvement. Curr Opin Rheumatol. 2018; 30(5):506–513. PMID: 29916847.
Article4. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26(5):589–595. PMID: 20080505.
Article5. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20(9):1297–1303. PMID: 20644199.
Article6. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016; 17(1):122. PMID: 27268795.
Article7. Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016; 37(3):235–241. PMID: 26555599.
Article8. Martinez-Quiles N, Goldbach-Mansky R. Updates on autoinflammatory diseases. Curr Opin Immunol. 2018; 55:97–105. PMID: 30453204.
Article9. Davidson S, Steiner A, Harapas CR, Masters SL. An update on autoinflammatory diseases: interferonopathies. Curr Rheumatol Rep. 2018; 20(7):38. PMID: 29846818.
Article10. Harapas CR, Steiner A, Davidson S, Masters SL. An update on autoinflammatory diseases: inflammasomopathies. Curr Rheumatol Rep. 2018; 20(7):40. PMID: 29846819.
Article11. Berteau F, Rouvière B, Nau A, Le Berre R, Sarrabay G, Touitou I, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis. 2019; 78(5):e35. PMID: 29549169.
Article12. Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wójtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011; 117(7):2227–2236. PMID: 21088135.13. Hövelmeyer N, Reissig S, Xuan NT, Adams-Quack P, Lukas D, Nikolaev A, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol. 2011; 41(3):595–601. PMID: 21341261.
Article14. Vanoni F, Lava SAG, Fossali EF, Cavalli R, Simonetti GD, Bianchetti MG, et al. Neonatal systemic lupus erythematosus syndrome: a comprehensive review. Clin Rev Allergy Immunol. 2017; 53(3):469–476. PMID: 29116459.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A20 Protects Against Arthritis by Regulation of the NLRP3 Inflammasome
- Multifocal Intracranial Stenosis and Thunderclap Headache in a Patient with Heterozygous MFAP5 Mutation for Familial Thoracic Aortic Aneurysm and Dissection
- Establishment of Leukemia Mouse Model Using Mouse-Derived A20 Leukemic Cells, and Detection of Tumor Cells in Bone Marrow
- Haploinsufficiency A20 misdiagnosed as PFAPA (periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis) syndrome with Kikuchi disease
- A Case of Familial Annular Erythema
