Genetic Studies in Human Prion Diseases
- Affiliations
-
- 1Korea Zoonosis Research Institute, Chonbuk National University, Jeonju, Korea.
- 2Ilsong Institute of Life Science, Hallym University, Anyang, Korea. yskim@hallym.ac.kr
- KMID: 1786101
- DOI: http://doi.org/10.3346/jkms.2014.29.5.623
Abstract
- Human prion diseases are fatal neurodegenerative disorders that are characterized by spongiform changes, astrogliosis, and the accumulation of an abnormal prion protein (PrP(Sc)). Approximately 10%-15% of human prion diseases are familial variants that are caused by pathogenic mutations in the prion protein gene (PRNP). Point mutations or the insertions of one or more copies of a 24 bp repeat are associated with familial human prion diseases including familial Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker syndrome, and fatal familial insomnia. These mutations vary significantly in frequency between countries. Here, we compare the frequency of PRNP mutations between European countries and East Asians. Associations between single nucleotide polymorphisms (SNPs) of several candidate genes including PRNP and CJD have been reported. The SNP of PRNP at codon 129 has been shown to be associated with sporadic, iatrogenic, and variant CJD. The SNPs of several genes other than PRNP have been showed contradictory results. Case-control studies and genome-wide association studies have also been performed to identify candidate genes correlated with variant and/or sporadic CJD. This review provides a general overview of the genetic mutations and polymorphisms that have been analyzed in association with human prion diseases to date.
Keyword
MeSH Terms
Figure
-
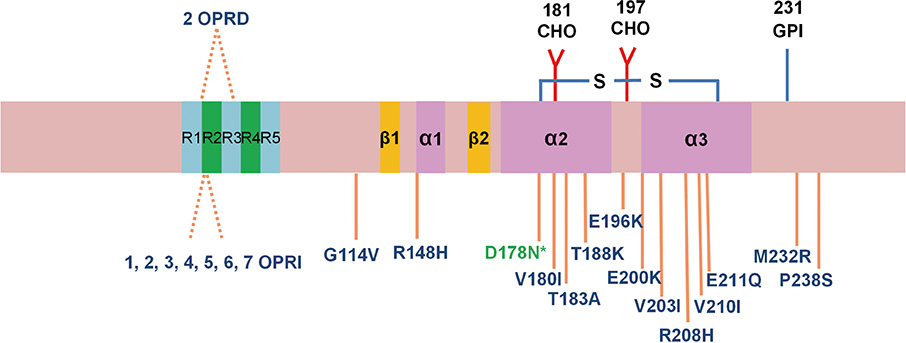
Fig. 1 Mutations in that PRNP gene the cause genetic Creutzfeldt-Jakob disease (CJD) or FFI in humans. D178N* is associated with familial CJD or fatal familial insomnia (FFI), depending on the allele present at codon 129 (Met, M = FFI, Val, V = familial CJD). The single-letter designations for the amino acids are as follows: D = aspartic acid, E = glutamic acid, G = glycine, H = histidine, I = isoleucine, K = lysine, M = methionine, N = asparagine, P = proline, Q = glutamine, R = arginine, S = serine, T = threonine, and V = valine. OPRI and OPRD indicate octapeptide repeat insertion and octapeptide repeat deletion, respectively. CHO, Asn-linked glycosylation sites; GPI, glycosylphosphatidylinositol.
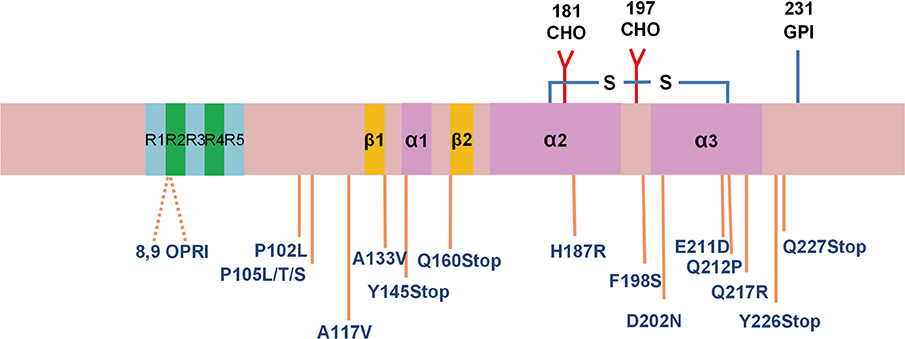
Fig. 2 Mutations in the PRNP gene that cause Gerstmann-Sträussler-Scheinker syndrome (GSS). The single-letter designations for the amino acids are as follows: A = alanine, D = aspartic acid, E = glutamic acid, F = phenylalanine, H = histidine, L = leucine, N = asparagine, P = proline, Q = glutamine, R = arginine, S = serine, T = threonine, V = valine, and Y = tyrosine. OPRI indicates the octapeptide repeat insertion. The stop indicates a stop codon. CHO, Asn-linked glycosylation sites; GPI, glycosylphosphatidylinositol.
Reference
-
1. Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998; 93:337–348.2. Brown K, Mastrianni JA. The prion diseases. J Geriatr Psychiatry Neurol. 2010; 23:277–298.3. Mastrianni JA. The genetics of prion diseases. Genet Med. 2010; 12:187–195.4. Kim HL, Do JY, Cho HJ, Jeon YC, Park SJ, Ma HI, Song JH, Lee Y, Choi H, Choi KC, et al. Dura mater graft-associated Creutzfeldt-Jakob disease: the first case in Korea. J Korean Med Sci. 2011; 26:1515–1517.5. Liao YC, Lebo RV, Clawson GA, Smuckler EA. Human prion protein cDNA: molecular cloning, chromosomal mapping, and biological implications. Science. 1986; 233:364–367.6. Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987; 51:229–240.7. Mead S. Prion disease genetics. Eur J Hum Genet. 2006; 14:273–281.8. Rodriguez MM, Peoc'h K, Haïk S, Bouchet C, Vernengo L, Mañana G, Salamano R, Carrasco L, Lenne M, Beaudry P, et al. A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology. 2005; 64:1455–1457.9. Nieto A, Goldfarb LG, Brown P, McCombie WR, Trapp S, Asher DM, Gajdusek DC. Codon 178 mutation in ethnically diverse Creutzfeldt-Jakob disease families. Lancet. 1991; 337:622–623.10. Kitamoto T, Ohta M, Doh-ura K, Hitoshi S, Terao Y, Tateishi J. Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun. 1993; 191:709–714.11. Nitrini R, Rosemberg S, Passos-Bueno MR, da Silva LS, Iughetti P, Papadopoulos M, Carrilho PM, Caramelli P, Albrecht S, Zatz M, et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann Neurol. 1997; 42:138–146.12. Finckh U, Müller-Thomsen T, Mann U, Eggers C, Marksteiner J, Meins W, Binetti G, Alberici A, Hock C, Nitsch RM, et al. High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet. 2000; 66:110–117.13. Peoc'h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Lauprêtre N, Laplanche JL. Identification of three novel mutations (E196K, V203I, E211Q) in the prion protein gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat. 2000; 15:482.14. Goldfarb LG, Korczyn AD, Brown P, Chapman J, Gajdusek DC. Mutation in codon 200 of scrapie amyloid precursor gene linked to Creutzfeldt-Jakob disease in Sephardic Jews of Libyan and non-Libyan origin. Lancet. 1990; 336:637–638.15. Mastrianni JA, Iannicola C, Myers RM, DeArmond S, Prusiner SB. Mutation of the prion protein gene at codon 208 in familial Creutzfeldt-Jakob disease. Neurology. 1996; 47:1305–1312.16. Pocchiari M, Salvatore M, Cutruzzolá F, Genuardi M, Allocatelli CT, Masullo C, Macchi G, Alemá G, Galgani S, Xi YG, et al. A new point mutation of the prion protein gene in Creutzfeldt-Jakob disease. Ann Neurol. 1993; 34:802–807.17. Hoque MZ, Kitamoto T, Furukawa H, Muramoto T, Tateishi J. Mutation in the prion protein gene at codon 232 in Japanese patients with Creutzfeldt-Jakob disease: a clinicopathological, immunohistochemical and transmission study. Acta Neuropathol. 1996; 92:441–446.18. Windl O, Giese A, Schulz-Schaeffer W, Zerr I, Skworc K, Arendt S, Oberdieck C, Bodemer M, Poser S, Kretzschmar HA. Molecular genetics of human prion diseases in Germany. Hum Genet. 1999; 105:244–252.19. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, Westaway D, Ott J, Prusiner SB. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989; 338:342–345.20. Kitamoto T, Amano N, Terao Y, Nakazato Y, Isshiki T, Mizutani T, Tateishi J. A new inherited prion disease (PrP-P105L mutation) showing spastic paraparesis. Ann Neurol. 1993; 34:808–813.21. Rogaeva E, Zadikoff C, Ponesse J, Schmitt-Ulms G, Kawarai T, Sato C, Salehi-Rad S, St George-Hyslop P, Lang AE. Childhood onset in familial prion disease with a novel mutation in the PRNP gene. Arch Neurol. 2006; 63:1016–1021.22. Tunnell E, Wollman R, Mallik S, Cortes CJ, Dearmond SJ, Mastrianni JA. A novel PRNP-P105S mutation associated with atypical prion disease and a rare PrPSc conformation. Neurology. 2008; 71:1431–1438.23. Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. Pro-leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun. 1989; 163:974–979.24. Panegyres PK, Toufexis K, Kakulas BA, Cernevakova L, Brown P, Ghetti B, Piccardo P, Dlouhy SR. A new PRNP mutation (G131V) associated with Gerstmann-Sträussler-Scheinker disease. Arch Neurol. 2001; 58:1899–1902.25. Kitamoto T, Iizuka R, Tateishi J. An amber mutation of prion protein in Gerstmann-Sträussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun. 1993; 192:525–531.26. Bütefisch CM, Gambetti P, Cervenakova L, Park KY, Hallett M, Goldfarb LG. Inherited prion encephalopathy associated with the novel PRNP H187R mutation: a clinical study. Neurology. 2000; 55:517–522.27. Hsiao K, Dlouhy SR, Farlow MR, Cass C, Da Costa M, Conneally PM, Hodes ME, Ghetti B, Prusiner SB. Mutant prion proteins in Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Nat Genet. 1992; 1:68–71.28. Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998; 57:979–988.29. Jansen C, Voet W, Head MW, Parchi P, Yull H, Verrips A, Wesseling P, Meulstee J, Baas F, van Gool WA, et al. A novel seven-octapeptide repeat insertion in the prion protein gene (PRNP) in a Dutch pedigree with Gerstmann-Sträussler-Scheinker disease phenotype: comparison with similar cases from the literature. Acta Neuropathol. 2011; 121:59–68.30. Medori R, Tritschler HJ, LeBlanc A, Villare F, Manetto V, Chen HY, Xue R, Leal S, Montagna P, Cortelli P, et al. Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med. 1992; 326:444–449.31. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003; 66:213–239.32. Liberski PP. Gerstmann-Sträussler-Scheinker disease. Adv Exp Med Biol. 2012; 724:128–137.33. Lugaresi E, Provini F. Fatal familial insomnia and agrypnia excitata. Rev Neurol Dis. 2007; 4:145–152.34. Lloyd S, Mead S, Collinge J. Genetics of prion disease. Top Curr Chem. 2011; 305:1–22.35. Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991; 352:340–342.36. Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1998; 43:826–828.37. Jeong BH, Lee KH, Kim NH, Jin JK, Kim JI, Carp RI, Kim YS. Association of sporadic Creutzfeldt-Jakob disease with homozygous genotypes at PRNP codons 129 and 219 in the Korean population. Neurogenetics. 2005; 6:229–232.38. Mead S, Poulter M, Uphill J, Beck J, Whitfield J, Webb TE, Campbell T, Adamson G, Deriziotis P, Tabrizi SJ, et al. Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol. 2009; 8:57–66.39. Sanchez-Juan P, Bishop MT, Aulchenko YS, Brandel JP, Rivadeneira F, Struchalin M, Lambert JC, Amouyel P, Combarros O, Sainz J, et al. Genome-wide study links MTMR7 gene to variant Creutzfeldt-Jakob risk. Neurobiol Aging. 2012; 33:1487.e21–1487.e28.40. Mead S, Uphill J, Beck J, Poulter M, Campbell T, Lowe J, Adamson G, Hummerich H, Klopp N, Rückert IM, et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet. 2012; 21:1897–1906.41. Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011; 121:21–37.42. Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005; 118:166–174.43. Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010; 133:3043–3057.44. Moe Lee S, Ran Ju Y, Choi BY, Wook Hyeon J, Sun Park J, Kyeong Kim C, Yeon Kim S. Genotype patterns and characteristics of PRNP in the Korean population. Prion. 2012; 6:375–382.45. Jeong BH, Jeon YC, Lee YJ, Cho HJ, Park SJ, Chung DI, Kim J, Kim SH, Kim HT, Choi EK, et al. Creutzfeldt-Jakob disease with the V203I mutation and M129V polymorphism of the prion protein gene (PRNP) and a 17 kDa prion protein fragment. Neuropathol Appl Neurobiol. 2010; 36:558–563.46. Choi BY, Kim SY, Seo SY, An SS, Kim S, Park SE, Lee SH, Choi YJ, Kim SJ, Kim CK, et al. Mutations at codons 178, 200-129, and 232 contributed to the inherited prion diseases in Korean patients. BMC Infect Dis. 2009; 9:132.47. Yang TI, Jung DS, Ahn BY, Jeong BH, Cho HJ, Kim YS, Na DL, Geschwind MD, Kim EJ. Familial Creutzfeldt-Jakob disease with V180I mutation. J Korean Med Sci. 2010; 25:1097–1100.48. Yeo MJ, Lee SH, Lee SY, Jeon YC, Park SJ, Cho HJ, Choi KC, Kim YS, Kim SH. Familial Creutzfeldt-Jakob disease with a mutation at codon 180 presenting with an atypical phenotype. J Clin Neurosci. 2013; 20:180–182.49. Jeong BH, Ju WK, Huh K, Lee EA, Choi IS, Im JH, Choi EK, Kim YS. Molecular analysis of prion protein gene (PRNP) in Korean patients with Creutzfeldt-Jakob disease. J Korean Med Sci. 1998; 13:234–240.50. Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011; 8:559.51. Park MJ, Jo HY, Cheon SM, Choi SS, Kim YS, Kim JW. A case of gerstmann-sträussler-scheinker disease. J Clin Neurol. 2010; 6:46–50.52. Giaccone G, Tagliavini F, Verga L, Frangione B, Farlow MR, Bugiani O, Ghetti B. Neurofibrillary tangles of the Indiana kindred of Gerstmann-Sträussler-Scheinker disease share antigenic determinants with those of Alzheimer disease. Brain Res. 1990; 530:325–329.53. Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J, Seiler C, Frangione B, et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci U S A. 1996; 93:744–748.54. Yamada M, Itoh Y, Inaba A, Wada Y, Takashima M, Satoh S, Kamata T, Okeda R, Kayano T, Suematsu N, et al. An inherited prion disease with a PrP P105L mutation: clinicopathologic and PrP heterogeneity. Neurology. 1999; 53:181–188.55. Jeong BH, Nam JH, Lee YJ, Lee KH, Jang MK, Carp RI, Lee HD, Ju YR, Ahn Jo S, Park KY, et al. Polymorphisms of the prion protein gene (PRNP) in a Korean population. J Hum Genet. 2004; 49:319–324.56. Doh-ura K, Kitamoto T, Sakaki Y, Tateishi J. CJD discrepancy. Nature. 1991; 353:801–802.57. Dermaut B, Croes EA, Rademakers R, Van den Broeck M, Cruts M, Hofman A, van Duijn CM, Van Broeckhoven C. PRNP Val129 homozygosity increases risk for early-onset Alzheimer's disease. Ann Neurol. 2003; 53:409–412.58. Jansen C, Parchi P, Capellari S, Ibrahim-Verbaas CA, Schuur M, Strammiello R, Corrado P, Bishop MT, van Gool WA, Verbeek MM, et al. Human prion diseases in the Netherlands (1998-2009): clinical, genetic and molecular aspects. PLoS One. 2012; 7:e36333.59. Calero O, Bullido MJ, Clarimón J, Frank-García A, Martínez-Martín P, Lleó A, Rey MJ, Rábano A, Blesa R, Gómez-Isla T, et al. Genetic cross-interaction between APOE and PRNP in sporadic Alzheimer's and Creutzfeldt-Jakob diseases. PLoS One. 2011; 6:e22090.60. Brandel JP, Preece M, Brown P, Croes E, Laplanche JL, Agid Y, Will R, Alpérovitch A. Distribution of codon 129 genotype in human growth hormone-treated CJD patients in France and the UK. Lancet. 2003; 362:128–130.61. Laplanche JL, Delasnerie-Lauprêtre N, Brandel JP, Chatelain J, Beaudry P, Alpérovitch A, Launay JM. Molecular genetics of prion diseases in France: French Research Group on Epidemiology of Human Spongiform Encephalopathies. Neurology. 1994; 44:2347–2351.62. Geldermann H, Bartenschlager H, Preuss S, Melchinger-Wild E, Herzog K, Zerr I. Polymorphic microsatellite sites in the PRNP region point to excess of homozygotes in Creutzfeldt-Jakob disease patients. Gene. 2006; 382:66–70.63. Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991; 337:1441–1442.64. Deslys JP, Marcé D, Dormont D. Similar genetic susceptibility in iatrogenic and sporadic Creutzfeldt-Jakob disease. J Gen Virol. 1994; 75:23–27.65. Ward HJ, Head MW, Will RG, Ironside JW. Variant Creutzfeldt-Jakob disease. Clin Lab Med. 2003; 23:87–108.66. Petraroli R, Pocchiari M. Codon 219 polymorphism of PRNP in healthy Caucasians and Creutzfeldt-Jakob disease patients. Am J Hum Genet. 1996; 58:888–889.67. Palmer MS, Mahal SP, Campbell TA, Hill AF, Sidle KC, Laplanche JL, Collinge J. Deletions in the prion protein gene are not associated with CJD. Hum Mol Genet. 1993; 2:541–544.68. Mead S, Mahal SP, Beck J, Campbell T, Farrall M, Fisher E, Collinge J. Sporadic - but not variant - Creutzfeldt-Jakob disease is associated with polymorphisms upstream of PRNP exon 1. Am J Hum Genet. 2001; 69:1225–1235.69. Vollmert C, Windl O, Xiang W, Rosenberger A, Zerr I, Wichmann HE, Bickeböller H, Illig T. KORA group, Kretzschmar HA. Significant association of a M129V independent polymorphism in the 5' UTR of the PRNP gene with sporadic Creutzfeldt-Jakob disease in a large German case-control study. J Med Genet. 2006; 43:e53.70. Jeong BH, Lee KH, Lee YJ, Kim YH, Cho YS, Carp RI, Kim YS. PRNP 1368 polymorphism is not associated with sporadic Creutzfeldt-Jakob disease in the Korean population. Eur J Neurol. 2008; 15:846–850.71. Croes EA, Alizadeh BZ, Bertoli-Avella AM, Rademaker T, Vergeer-Drop J, Dermaut B, Houwing-Duistermaat JJ, Wientjens DP, Hofman A, Van Broeckhoven C, et al. Polymorphisms in the prion protein gene and in the doppel gene increase susceptibility for Creutzfeldt-Jakob disease. Eur J Hum Genet. 2004; 12:389–394.72. Bratosiewicz-Wąsik J, Smoleń-Dzirba J, Rozemuller AJ, Jansen C, Spliet W, Jansen GH, Wąsik TJ, Liberski PP. Association between the PRNP 1368 polymorphism and the occurrence of sporadic Creutzfeldt-Jakob disease. Prion. 2012; 6:413–416.73. Sanchez-Juan P, Bishop MT, Croes EA, Knight RS, Will RG, van Duijn CM, Manson JC. A polymorphism in the regulatory region of PRNP is associated with increased risk of sporadic Creutzfeldt-Jakob disease. BMC Med Genet. 2011; 12:73.74. Bratosiewicz-Wasik J, Liberski PP, Golanska E, Jansen GH, Wasik TJ. Regulatory sequences of the PRNP gene influence susceptibility to sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 2007; 411:163–167.75. McCormack JE, Baybutt HN, Everington D, Will RG, Ironside JW, Manson JC. PRNP contains both intronic and upstream regulatory regions that may influence susceptibility to Creutzfeldt-Jakob Disease. Gene. 2002; 288:139–146.76. Lu K, Wang W, Xie Z, Wong BS, Li R, Petersen RB, Sy MS, Chen SG. Expression and structural characterization of the recombinant human doppel protein. Biochemistry. 2000; 39:13575–13583.77. Mead S, Beck J, Dickinson A, Fisher EM, Collinge J. Examination of the human prion protein-like gene doppel for genetic susceptibility to sporadic and variant Creutzfeldt-Jakob disease. Neurosci Lett. 2000; 290:117–120.78. Peoc'h K, Guérin C, Brandel JP, Launay JM, Laplanche JL. First report of polymorphisms in the prion-like protein gene (PRND): implications for human prion diseases. Neurosci Lett. 2000; 286:144–148.79. Schröder B, Franz B, Hempfling P, Selbert M, Jürgens T, Kretzschmar HA, Bodemer M, Poser S, Zerr I. Polymorphisms within the prion-like protein gene (Prnd) and their implications in human prion diseases, Alzheimer's disease and other neurological disorders. Hum Genet. 2001; 109:319–325.80. Jeong BH, Kim NH, Kim JI, Carp RI, Kim YS. Polymorphisms at codons 56 and 174 of the prion-like protein gene (PRND) are not associated with sporadic Creutzfeldt-Jakob disease. J Hum Genet. 2005; 50:311–314.81. Jeong BH, Kim NH, Choi EK, Lee C, Song YH, Kim JI, Carp RI, Kim YS. Polymorphism at 3' UTR +28 of the prion-like protein gene is associated with sporadic Creutzfeldt-Jakob disease. Eur J Hum Genet. 2005; 13:1094–1097.82. Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene. 2003; 314:89–102.83. Beck JA, Campbell TA, Adamson G, Poulter M, Uphill JB, Molou E, Collinge J, Mead S. Association of a null allele of SPRN with variant Creutzfeldt-Jakob disease. J Med Genet. 2008; 45:813–817.84. Augereau P, Garcia M, Mattei MG, Cavailles V, Depadova F, Derocq D, Capony F, Ferrara P, Rochefort H. Cloning and sequencing of the 52K cathepsin D complementary deoxyribonucleic acid of MCF7 breast cancer cells and mapping on chromosome 11. Mol Endocrinol. 1988; 2:186–192.85. Kovács GG, Gelpi E, Ströbel T, Ricken G, Nyengaard JR, Bernheimer H, Budka H. Involvement of the endosomal-lysosomal system correlates with regional pathology in Creutzfeldt-Jakob disease. J Neuropathol Exp Neurol. 2007; 66:628–636.86. Bishop MT, Kovacs GG, Sanchez-Juan P, Knight RS. Cathepsin D SNP associated with increased risk of variant Creutzfeldt-Jakob disease. BMC Med Genet. 2008; 9:31.87. Jeong BH, Lee KH, Lee YJ, Yun J, Park YJ, Bae Y, Kim YH, Cho YS, Choi EK, Carp RI, et al. Genetic association of a cathepsin D polymorphism and sporadic Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord. 2009; 28:302–306.88. Kovacs GG, Sanchez-Juan P, Ströbel T, Schuur M, Poleggi A, Nocentini S, Giannattasio C, Belay G, Bishop M, Capellari S, et al. Cathepsin D (C224T) polymorphism in sporadic and genetic Creutzfeldt-Jakob disease. Alzheimer Dis Assoc Disord. 2010; 24:104–107.89. Lloyd SE, Maytham EG, Pota H, Grizenkova J, Molou E, Uphill J, Hummerich H, Whitfield J, Alpers MP, Mead S, et al. HECTD2 is associated with susceptibility to mouse and human prion disease. PLoS Genet. 2009; 5:e1000383.90. Jeong BH, Lee KH, Lee YJ, Yun J, Park YJ, Cho HJ, Kim YH, Cho YS, Choi EK, Carp RI, et al. Absence of association between two HECTD2 polymorphisms and sporadic Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord. 2011; 31:146–151.91. Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, et al. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet. 2008; 40:1076–1083.92. Caffrey TM, Wade-Martins R. Functional MAPT haplotypes: bridging the gap between genotype and neuropathology. Neurobiol Dis. 2007; 27:1–10.93. Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I. Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 2009; 118:53–69.94. Sánchez-Juan P, Bishop MT, Green A, Giannattasio C, Arias-Vasquez A, Poleggi A, Knight RS, van Duijn CM. No evidence for association between tau gene haplotypic variants and susceptibility to Creutzfeldt-Jakob disease. BMC Med Genet. 2007; 8:77.95. Das HK, McPherson J, Bruns GA, Karathanasis SK, Breslow JL. Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J Biol Chem. 1985; 260:6240–6247.96. Martin ER, Lai EH, Gilbert JR, Rogala AR, Afshari AJ, Riley J, Finch KL, Stevens JF, Livak KJ, Slotterbeck BD, et al. SNPing away at complex diseases: analysis of single-nucleotide polymorphisms around APOE in Alzheimer disease. Am J Hum Genet. 2000; 67:383–394.97. Amouyel P, Vidal O, Launay JM, Laplanche JL. The apolipoprotein E alleles as major susceptibility factors for Creutzfeldt-Jakob disease: the French Research Group on Epidemiology of Human Spongiform Encephalopathies. Lancet. 1994; 344:1315–1318.98. Van Everbroeck B, Croes EA, Pals P, Dermaut B, Jansen G, van Duijn CM, Cruts M, Van Broeckhoven C, Martin JJ, Cras P. Influence of the prion protein and the apolipoprotein E genotype on the Creutzfeldt-Jakob Disease phenotype. Neurosci Lett. 2001; 313:69–72.99. Nakagawa Y, Kitamoto T, Furukawa H, Ogomori K, Tateishi J. Allelic variation of apolipoprotein E in Japanese sporadic Creutzfeldt-Jakob disease patients. Neurosci Lett. 1995; 187:209–211.100. Pickering-Brown SM, Mann DM, Owen F, Ironside JW, de Silva R, Roberts DA, Balderson DJ, Cooper PN. Allelic variations in apolipoprotein E and prion protein genotype related to plaque formation and age of onset in sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 1995; 187:127–129.101. Mullan M, Houlden H, Windelspecht M, Fidani L, Lombardi C, Diaz P, Rossor M, Crook R, Hardy J, Duff K, et al. A locus for familial early-onset Alzheimer's disease on the long arm of chromosome 14, proximal to the alpha 1-antichymotrypsin gene. Nat Genet. 1992; 2:340–342.102. Salvatore M, Seeber AC, Nacmias B, Petraroli R, Sorbi S, Pocchiari M. Alpha1 antichymotrypsin signal peptide polymorphism in sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 1997; 227:140–142.103. Yamazaki K, Mizui Y, Tanaka I. Radiation hybrid mapping of human ADAM10 gene to chromosome 15. Genomics. 1997; 45:457–459.104. Laffont-Proust I, Faucheux BA, Hässig R, Sazdovitch V, Simon S, Grassi J, Hauw JJ, Moya KL, Haïk S. The N-terminal cleavage of cellular prion protein in the human brain. FEBS Lett. 2005; 579:6333–6337.105. Plamont MA, Chasseigneaux S, Delasnerie-Lauprêtre N, Beaudry P, Peoc'h K, Laplanche JL. Variation at the ADAM10 gene locus is not associated with Creutzfeldt-Jakob disease. Neurosci Lett. 2003; 344:132–134.106. Gauczynski S, Peyrin JM, Haïk S, Leucht C, Hundt C, Rieger R, Krasemann S, Deslys JP, Dormont D, Lasmézas CI, et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001; 20:5863–5875.107. Rieger R, Edenhofer F, Lasmézas CI, Weiss S. The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med. 1997; 3:1383–1388.108. Yun J, Jin HT, Lee YJ, Choi EK, Carp RI, Jeong BH, Kim YS. The first report of RPSA polymorphisms, also called 37/67 kDa LRP/LR gene, in sporadic Creutzfeldt-Jakob disease (CJD). BMC Med Genet. 2011; 12:108.109. Muratake T, Hayashi S, Ichikawa T, Kumanishi T, Ichimura Y, Kuwano R, Isobe T, Wang Y, Minoshima S, Shimizu N, et al. Structural organization and chromosomal assignment of the human 14-3-3 eta chain gene (YWHAH). Genomics. 1996; 36:63–69.110. Stoeck K, Sanchez-Juan P, Gawinecka J, Green A, Ladogana A, Pocchiari M, Sanchez-Valle R, Mitrova E, Sklaviadis T, Kulczycki J, et al. Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain. 2012; 135:3051–3061.111. Yun J, Jeong BH, Kim HJ, Park YJ, Lee YJ, Choi EK, Carp RI, Kim YS. A polymorphism in the YWHAH gene encoding 14-3-3 eta that is not associated with sporadic Creutzfeldt-Jakob disease (CJD). Mol Biol Rep. 2012; 39:3619–3625.112. Tommerup N, Leffers H. Assignment of the human genes encoding 14, 3-3 Eta (YWHAH) to 22q12, 14-3-3 zeta (YWHAZ) to 2p25.1-p25.2, and 14-3-3 beta (YWHAB) to 20q13.1 by in situ hybridization. Genomics. 1996; 33:149–150.113. Mei GY, Li Y, Wang GR, Zhang BY, Tian C, Chen C, Zhou RM, Wang X, Li XL, Wang KX, et al. Molecular interaction between PrP protein and the signal protein 14-3-3 beta. Bing Du Xue Bao. 2009; 25:208–212.114. Jeong BH, Jin HT, Choi EK, Carp RI, Kim YS. Lack of association between 14-3-3 beta gene (YWHAB) polymorphisms and sporadic Creutzfeldt-Jakob disease (CJD). Mol Biol Rep. 2012; 39:10647–10653.115. Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004; 18:1034–1036.116. Calero O, Bullido MJ, Clarimón J, Frank-García A, Martínez-Martín P, Lleó A, Rey MJ, Sastre I, Rábano A, de Pedro-Cuesta J, et al. A common BACE1 polymorphism is a risk factor for sporadic Creutzfeldt-Jakob disease. PLoS One. 2012; 7:e43926.117. Dreses-Werringloer U, Lambert JC, Vingtdeux V, Zhao H, Vais H, Siebert A, Jain A, Koppel J, Rovelet-Lecrux A, Hannequin D, et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer's disease risk. Cell. 2008; 133:1149–1161.118. Calero O, Bullido MJ, Clarimón J, Hortigüela R, Frank-García A, Martínez-Martín P, Lleó A, Rey MJ, Sastre I, Rábano A, et al. Genetic variability of the gene cluster CALHM 1-3 in sporadic Creutzfeldt-Jakob disease. Prion. 2012; 6:407–412.119. Jeong BH, Kim YS. Creutzfeldt-Jakob disease susceptibility: an approach to discovering multiple candidate genes for human prion diseases. Adv Genet Eng Biotechnol. 2012; 1:2.120. Jeong BH, Kim HJ, Lee KH, Carp RI, Kim YS. RARB and STMN2 polymorphisms are not associated with sporadic Creutzfeldt-Jakob disease (CJD) in the Korean population. Mol Biol Rep. 2014; 41:2389–2395.
