Ann Pediatr Endocrinol Metab.
2022 Dec;27(4):247-255. 10.6065/apem.2244120.060.
Growth plate extracellular matrix defects and short stature in children
- Affiliations
-
- 1Department of Pediatrics, University of Chieti, Chieti, Italy
- KMID: 2537236
- DOI: http://doi.org/10.6065/apem.2244120.060
Abstract
- Many etiological factors causing short stature have already been identified in humans. In the last few years, the advent of new techniques for the detection of chromosomal and molecular abnormalities has made it possible to better identify patients with genetic causes of growth failure. Some of these factors directly affect the development and growth of the skeleton, since they damage the epiphyseal growth plate, where linear growth occurs, influencing chondrogenesis. In particular, defects in genes involved in the organization and function of the growth plate are responsible for several well-known conditions with short stature. These genes play a pivotal role in various mechanisms involving the extracellular matrix, intracellular signaling, paracrine signaling, endocrine signaling, and epigenetic regulation. In this review, we will discuss the genes involved in extracellular matrix disorders. The identification of genetic defects in linear growth failure is important for clinicians and researchers in order to improve the care of children affected by growth disorders.
Keyword
Figure
-
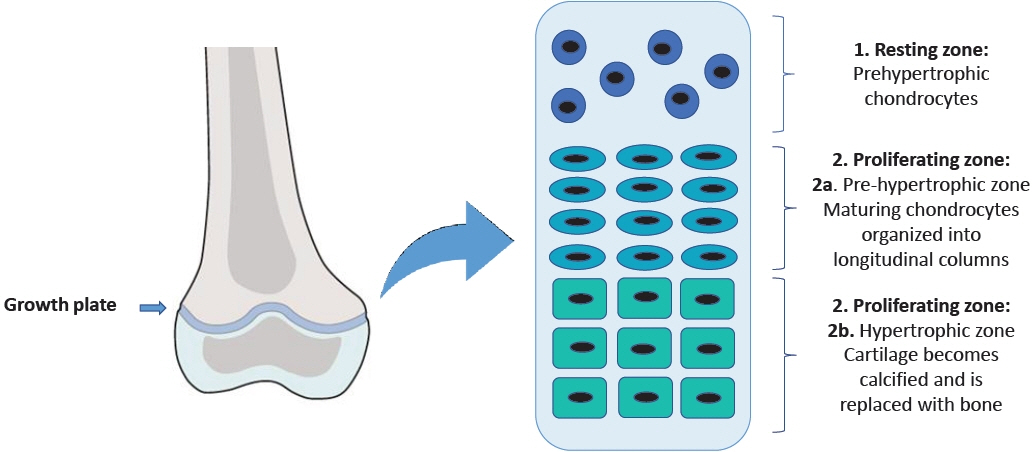
Fig. 1. The structure of the growth plate.
Reference
-
References
1. Bang P. Statement 3: a low serum IGF-I level in idiopathic short stature patients indicates partial GH insensitivity. Pediatr Endocrinol Rev. 2008; 5 Suppl 3:841–6.2. Patel R, Dave C, Agarwal N, Mendpara H, Shukla R, Bajpai A. Predictive value of IAP 2015, IAP 2007 and WHO growth charts in identifying pathological short stature. Indian Pediatr. 2021; 58:149–51.3. Chiarelli F, Primavera M, Mastromauro C. Evaluation and management of a child with short stature. Minerva Pediatr. 2020; 72:452–61.4. Yue S, Whalen P, Jee YH. Genetic regulation of linear growth. Ann Pediatr Endocrinol Metab. 2019; 24:2–14.5. Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res. 2005; 64:157–65.6. Chen H, Tan XN, Hu S, Liu RQ, Peng LH, Li YM, et al. Molecular mechanisms of chondrocyte proliferation and differentiation. Front Cell Dev Biol. 2021; 9:664168.7. Tsang KY, Tsang SW, Chan D, Cheah KS. The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Defects Res C Embryo Today. 2014; 102:52–73.8. Faienza MF, Chiarito M, Brunetti G, D'Amato G. Growth plate gene involvement and isolated short stature. Endocrine. 2021; 71:28–34.9. Schwartz NB, Domowicz M. Chondrodysplasias due to proteoglycan defects. Glycobiology. 2002; 12:57R–68R.10. Cortes M, Baria AT, Schwartz NB. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development. 2009; 136:1697–706.11. Rellmann Y, Dreier R. Different forms of ER stress in chondrocytes result in short stature disorders and degenerative cartilage diseases: new insights by cartilagespecific ERp57 knockout mice. Oxid Med Cell Longev. 2018; 2018:8421394.12. Patterson SE, Dealy CN. Mechanisms and models of endoplasmic reticulum stress in chondrodysplasia. Dev Dyn. 2014; 243:875–93.13. Krakow D. Skeletal dysplasias. Clin Perinatol. 2015; 42:301–19.14. Krakow D, Rimoin DL. The skeletal dysplasias. Genet Med. 2010; 12:327–41.15. Myllyharju J, Kivirikko KI. Collagens and collagen-related diseases. Ann Med. 2001; 33:7–21.16. Lian C, Wang X, Qiu X, Wu Z, Gao B, Liu L, et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1-SMAD1 interaction. Bone Res. 2019; 7:8.17. Antipova O, Orgel JP. In situ D-periodic molecular structure of type II collagen. J Biol Chem. 2010; 285:7087–96.18. Prein C, Warmbold N, Farkas Z, Schieker M, Aszodi A, Clausen-Schaumann H. Structural and mechanical properties of the proliferative zone of the developing murine growth plate cartilage assessed by atomic force microscopy. Matrix Biol. 2016; 50:1–15.19. Chung HJ, Jensen DA, Gawron K, Steplewski A, Fertala A. R992C (p.R1192C) Substitution in collagen II alters the structure of mutant molecules and induces the unfolded protein response. J Mol Biol. 2009; 390:306–18.20. Rajpar MH, McDermott B, Kung L, Eardley R, Knowles L, Heeran M, et al. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet. 2009; 5:e1000691.21. Bateman JF, Freddi S, Nattrass G, Savarirayan R. Tissuespecific RNA surveillance? Nonsense-mediated mRNA decay causes collagen X haploinsufficiency in Schmid metaphyseal chondrodysplasia cartilage. Hum Mol Genet. 2003; 12:217–25.22. Pihlajamaa T, Vuoristo MM, Annunen S, Perälä M, Prockop DJ, Ala-Kokko L. Human COL9A1 and COL9A2 genes. Two genes of 90 and 15 kb code for similar polypeptides of the same collagen molecule. Matrix Biol. 1998; 17:237–41.23. Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perälä M, Carter L, et al. A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am J Hum Genet. 2001; 69:969–80.24. Unger S, Bonafé L, Superti-Furga A. Multiple epiphyseal dysplasia: clinical and radiographic features, differential diagnosis and molecular basis. Best Pract Res Clin Rheumatol. 2008; 22:19–32.25. Haga N, Nakamura K, Takikawa K, Manabe N, Ikegawa S, Kimizuka M. Stature and severity in multiple epiphyseal dysplasia. J Pediatr Orthop. 1998; 18:394–7.26. Plumb DA, Ferrara L, Torbica T, Knowles L, Mironov A Jr, Kadler KE, et al. Collagen XXVII organises the pericellular matrix in the growth plate. PLoS One. 2011; 6:e29422.27. Hjorten R, Hansen U, Underwood RA, Telfer HE, Fernandes RJ, Krakow D, et al. Type XXVII collagen at the transition of cartilage to bone during skeletogenesis. Bone. 2007; 41:535–42.28. Li Y, Lacerda DA, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995; 80:423–30.29. Van Camp G, Snoeckx RL, Hilgert N, van den Ende J, Fukuoka H, Wagatsuma M, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet. 2006; 79:449–57.30. Annunen S, Körkkö J, Czarny M, Warman ML, Brunner HG, Kääriäinen H, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999; 65:974–83.31. Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH, Warman ML. Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am J Hum Genet. 1998; 62:816–23.32. Tompson SW, Bacino CA, Safina NP, Bober MB, Proud VK, Funari T, et al. Fibrochondrogenesis results from mutations in the COL11A1 type XI collagen gene. Am J Hum Genet. 2010; 87:708–12.33. Pace JM, Corrado M, Missero C, Byers PH. Identification, characterization and expression analysis of a new fibrillar collagen gene, COL27A1. Matrix Biol. 2003; 22:3–14.34. Kotabagi S, Shah H, Shukla A, Girisha KM. Second family provides further evidence for causation of Steel syndrome by biallelic mutations in COL27A1. Clin Genet. 2017; 92:323–26.35. Gariballa N, Ben-Mahmoud A, Komara M, Al-Shamsi AM, John A, Ali BR, et al. A novel aberrant splice site mutation in COL27A1 is responsible for Steel syndrome and extension of the phenotype to include hearing loss. Am J Med Genet A. 2017; 173:1257–63.36. Evie Kritioti, Athina Theodosiou, Nayia Nicolaou, Angelos Alexandrou, Ioannis Papaevripidou, Elisavet Efstathiou, et al. First reported case of Steel syndrome in the European population: a novel homozygous mutation in COL27A1 and review of the literature. Eur J Med Genet. 2020; 63:103939.37. Sakai LY, Keene DR, Renard M, De Backer J. FBN1: the disease-causing gene for Marfan syndrome and other genetic disorders. Gene. 2016; 591:279–91.38. Sakai LY, Keene DR. Fibrillin protein pleiotropy: acromelic dysplasias. Matrix Biol. 2019; 80:6–13.39. Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-β-binding proteins. Matrix Biol. 2015; 47:44–53.40. Cecchi A, Ogawa N, Martinez HR, Carlson A, Fan Y, Penny DJ, et al. Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome. Am J Med Genet A. 2013; 161A:2305–10.41. Cook JR, Ramirez F. Clinical, diagnostic, and therapeutic aspects of the Marfan syndrome. Adv Exp Med Biol. 2014; 802:77–94.42. Le Goff C, Cormier-Daire V. From tall to short: the role of TGFβ signaling in growth and its disorders. Am J Med Genet C Semin Med Genet. 2012; 160C:145–53.43. Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, et al. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003; 40:34–6.44. Le Goff C, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, et al. Mutations in the TGFβ binding-proteinlike domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011; 89:7–14.45. Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL, et al. Microenvironmental regulation by fibrillin-1. PLoS Genet. 2012; 8:e1002425.46. Al Motawa MNA, Al Shehri MSS, Al Buali MJ, Al Agnam AAM. Weill-Marchesani syndrome, a rare presentation of severe short stature with review of the literature. Am J Case Rep. 2021; 22:e930824.47. Marzin P, Cormier-Daire V, Tsilou E. Weill-Marchesani Syndrome. 2007 Nov 1 [updated 2020 Dec 10]. In : Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle;1993-2022.48. Allali S, Le Goff C, Pressac-Diebold I, Pfennig G, Mahaut C, Dagoneau N, et al. Molecular screening of ADAMTSL2 gene in 33 patients reveals the genetic heterogeneity of geleophysic dysplasia. J Med Genet. 2011; 48:417–21.49. Globa E, Zelinska N, Dauber A. The clinical cases of geleophysic dysplasia: one gene, different phenotypes. Case Rep Endocrinol. 2018; 2018:8212417.50. Kochhar A, Kirmani S, Cetta F, Younge B, Hyland JC, Michels V. Similarity of geleophysic dysplasia and weill-marchesani syndrome. Am J Med Genet A. 2013; 161A:3130–2.51. Legare JM, Modaff P, Strom SP, Pauli RM, Bartlett HL. Geleophysic dysplasia: 48 year clinical update with emphasis on cardiac care. Am J Med Genet A. 2018; 176:2237–42.52. Roughley PJ, Mort JS. The role of aggrecan in normal and osteoarthritic cartilage. J Exp Orthop. 2014; 1:8.53. Hauer NN, Sticht H, Boppudi S, Büttner C, Kraus C, Trautmann U, et al. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci Rep. 2017; 7:12225.54. Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J Clin Endocrinol Metab. 2017; 102:460–9.55. Gibson BG, Briggs MD. The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J Rare Dis. 2016; 11:86.56. Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet. 2009; 84:72–9.57. Sentchordi-Montané L, Aza-Carmona M, Benito-Sanz S, Barreda-Bonis AC, Sánchez-Garre C, Prieto-Matos P, et al. Heterozygous aggrecan variants are associated with short stature and brachydactyly: description of 16 probands and a review of the literature. Clin Endocrinol (Oxf ). 2018; 88:820–9.58. Anderson IJ, Tsipouras P, Scher C, Ramesar RS, Martell RW, Beighton P. Spondyloepiphyseal dysplasia, mild autosomal dominant type is not due to primary defects of type II collagen. Am J Med Genet. 1990; 37:272–6.59. Gleghorn L, Ramesar R, Beighton P, Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet. 2005; 77:484–90.60. Posey KL, Hecht JT. The role of cartilage oligomeric matrix protein (COMP) in skeletal disease. Curr Drug Targets. 2008; 9:869–77.61. Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002; 14:608–16.62. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. 2011; 3:a009712.63. Xu K, Zhang Y, Ilalov K, Carlson CS, Feng JQ, di Cesare PE, et al. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J Biol Chem. 2007; 282:11347–55.64. Posey KL, Coustry F, Hecht JT. Cartilage oligomeric matrix protein: COMPopathies and beyond. Matrix Biology. 2018; 71-72:161–73.65. Müller G, Michel A, Altenburg E. COMP (Cartilage Oligomeric Matrix Protein) is synthesized in ligament, tendon, meniscus, and articular cartilage. Connect Tissue Res. 1998; 39:233–44.66. Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: new etiologic developments. Am J Med Genet. 2001; 106:244–50.67. Briggs MD, Brock J, Ramsden SC, Bell PA. Genotype to phenotype correlations in cartilage oligomeric matrix protein associated chondrodysplasias. Eur J Hum Genet. 2014; 22:1278–82.68. Jackson GC, Mittaz-Crettol L, Taylor JA, Mortier GR, Spranger J, Zabel B, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution. Hum Mutat. 2012; 33:144–57.69. Posey KL, Hayes E, Haynes R, Hecht JT. Role of TSP-5/COMP in pseudoachondroplasia. Int J Biochem Cell Biol. 2004; 36:1005–12.70. Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019; 179:2393–419.71. Gamble C, Nguyen J, Hashmi SS, Hecht JT. Pseudoachondroplasia and painful sequelae. Am J Med Genet A. 2015; 167A:2618–22.72. Briggs MD, Wright MJ, Mortier GR. Multiple epiphyseal dysplasia, autosomal dominant. 2003 Jan 8 [updated 2019 Apr 25]. In : Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle;1993-2022.73. Klatt AR, Becker AKA, Neacsu CD, Paulsson M, Wagener R. The matrilins: modulators of extracellular matrix assembly. Int J Biochem Cell Biol. 2011; 43:320–30.74. Klatt AR, Nitsche DP, Kobbe B, Mörgelin M, Paulsson M, Wagener R. Molecular structure and tissue distribution of matrilin-3, a filament-forming extracellular matrix protein expressed during skeletal development. J Biol Chem. 2000; 275:3999–4006.75. Wagener R, Ehlen HWA, Ko YP, Kobbe B, Mann HH, Sengle G, et al. The matrilins - adaptor proteins in the extracellular matrix. FEBS Lett. 2005; 579:3323–9.76. Pei M, Luo J, Chen Q. Enhancing and maintaining chondrogenesis of synovial fibroblasts by cartilage extracellular matrix protein matrilins. Osteoarthritis Cartilage. 2008; 16:1110–7.77. Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat. 2002; 19:465–78.78. Posey KL, Yang Y, Veerisetty AC, Sharan SK, Hecht JT. Thrombospondins: from structure to therapeutics. Cell Mol Life Sci. 2008; 65:669–71.79. Nundlall S, Rajpar MH, Bell PA, Clowes C, Zeeff LAH, Gardner B, et al. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones. 2010; 15:835–49.80. Leighton MP, Nundlall S, Starborg T, Meadows RS, Suleman F, Knowles L, et al. Decreased chondrocyte proliferation and dysregulated apoptosis in the cartilage growth plate are key features of a murine model of epiphyseal dysplasia caused by a matn3 mutation. Hum Mol Genet. 2007; 16:1728–41.81. Wang LW, Kutz WE, Mead TJ, Beene LC, Singh S, Jenkins MW, et al. Adamts10 inactivation in mice leads to persistence of ocular microfibrils subsequent to reduced fibrillin-2 cleavage. Matrix Biol. 2019; 77:117–28.82. Mularczyk EJ, Singh M, Godwin ARF, Galli F, Humphreys N, Adamson AD, et al. ADAMTS10-mediated tissue disruption in Weill–Marchesani syndrome. Hum Mol Genet. 2018; 27:3675–87.83. Apte SS. A disintegrin-like and metalloprotease (reprolysintype) with thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009; 284:31493–7.84. Faivre L, Dollfus H, Lyonnet S, Alembik Y, Mégarbané A, Samples J, et al. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet A. 2003; 123A:204–7.85. Khan AO, Aldahmesh MA, Al-Ghadeer H, Mohamed JY, Alkuraya FS. Familial spherophakia with short stature caused by a novel homozygous ADAMTS17 mutation. Ophthalmic Genet. 2012; 33:235–9.86. Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, et al. Many sequence variants affecting diversity of adult human height. Nat Genet. 2008; 40:609–15.87. Hyytiäinen M, Taipale J, Heldin CH, Keski-Oja J. Recombinant latent transforming growth factor β-binding protein 2 assembles to fibroblast extracellular matrix and is susceptible to proteolytic processing and release. J Biol Chem. 1998; 273:20669–76.88. Parsi MK, Adams JRJ, Whitelock J, Gibson MA. LTBP-2 has multiple heparin/heparan sulfate binding sites. Matrix Biol. 2010; 29:393–401.89. Lewis CJ, Hedberg-Buenz A, DeLuca AP, Stone EM, Alward WLM, Fingert JH. Primary congenital and developmental glaucomas. Hum Mol Genet. 2017; 26(R1):R28–36.90. Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, et al. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012; 33:1182–7.91. Cadoff EB, Sheffer R, Wientroub S, Ovadia D, Meiner V, Schwarzbauer JE. Mechanistic insights into the cellular effects of a novel FN1 variant associated with a spondylometaphyseal dysplasia. Clin Genet. 2018; 94:429–37.92. Costantini A, Valta H, Baratang NV, Yap P, Bertola DR, Yamamoto GL, et al. Novel fibronectin mutations and expansion of the phenotype in spondylometaphyseal dysplasia with “corner fractures. ” Bone. 2019; 121:163–71.93. Lee CS, Fu H, Baratang N, Rousseau J, Kumra H, Sutton VR, et al. Mutations in fibronectin cause a subtype of spondylometaphyseal dysplasia with “corner fractures. ” Am J Hum Genet. 2017; 101:815–23.94. England J, McFarquhar A, Campeau PM. Spondylometaphyseal dysplasia, corner fracture type. 2020. In : Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle;1993-2022.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Novel genetic cause of idiopathic short stature
- Commentary on “Growth plate extracellular matrix defects and short stature in children”
- A new era of genetic diagnosis for short stature children: A review
- Genetic regulation of linear growth
- Identification of a novel heterozygous mutation of ACAN in a Korean family with proportionate short stature
