Apolipoprotein A1 Inhibits TGF-β1-Induced Epithelial-to-Mesenchymal Transition of Alveolar Epithelial Cells
- Affiliations
-
- 1Division of Allergy and Respiratory Medicine, Department of Internal Medicine, Soonchunhyang University Bucheon Hospital, Bucheon, Korea. swpark@schmc.ac.kr
- 2Department of Pathology, Soonchunhyang University Bucheon Hospital, Bucheon, Korea.
- 3Division of Allergy and Respiratory Medicine, Department of Internal Medicine, Soonchunhyang University Hospital, Seoul, Korea.
- 4Division of Allergy and Respiratory Medicine, Department of Internal Medicine, Soonchunhyang University Cheonan Hospital, Cheonan, Korea.
- KMID: 2326682
- DOI: http://doi.org/10.4046/trd.2016.79.3.143
Abstract
- BACKGROUND
Idiopathic pulmonary fibrosis (IPF) is a progressive and lethal lung disease characterized by the accumulation of excessive fibroblasts and myofibroblasts in the extracellular matrix. The transforming growth factor β1 (TGF-β1)-induced epithelial-to-mesenchymal transition (EMT) is thought to be a possible source of fibroblasts/myofibroblasts in IPF lungs. We have previously reported that apolipoprotein A1 (ApoA1) has anti-fibrotic activity in experimental lung fibrosis. In this study, we determine whether ApoA1 modulates TGF-β1-induced EMT in experimental lung fibrosis and clarify its mechanism of action.
METHODS
The A549 alveolar epithelial cell line was treated with TGF-β1 with or without ApoA1. Morphological changes and expression of EMT-related markers, including E-cadherin, N-cadherin, and α-smooth muscle actin were evaluated. Expressions of Smad and non-Smad mediators and TGF-β1 receptor type 1 (TβRI) and type 2 (TβRII) were measured. The silica-induced lung fibrosis model was established using ApoA1 overexpressing transgenic mice.
RESULTS
TGF-β1-treated A549 cells were changed to the mesenchymal morphology with less E-cadherin and more N-cadherin expression. The addition of ApoA1 inhibited the TGF-β1-induced change of the EMT phenotype. ApoA1 inhibited the TGF-β1-induced increase in the phosphorylation of Smad2 and 3 as well as that of ERK and p38 mitogen-activated protein kinase mediators. In addition, ApoA1 reduced the TGF-β1-induced increase in TβRI and TβRII expression. In a mouse model of silica-induced lung fibrosis, ApoA1 overexpression reduced the silica-mediated effects, which were increased N-cadherin and decreased E-cadherin expression in the alveolar epithelium.
CONCLUSION
Our data demonstrate that ApoA1 inhibits TGF-β1-induced EMT in experimental lung fibrosis.
Keyword
MeSH Terms
-
Actins
Animals
Apolipoprotein A-I*
Apolipoproteins*
Cadherins
Epithelial Cells*
Epithelial-Mesenchymal Transition
Epithelium
Extracellular Matrix
Fibroblasts
Fibrosis
Idiopathic Pulmonary Fibrosis
Lung
Lung Diseases
Mice
Mice, Transgenic
Myofibroblasts
Phenotype
Phosphorylation
Protein Kinases
Pulmonary Fibrosis
Transforming Growth Factor beta1
Transforming Growth Factors
Actins
Apolipoprotein A-I
Apolipoproteins
Cadherins
Protein Kinases
Transforming Growth Factor beta1
Transforming Growth Factors
Figure
-
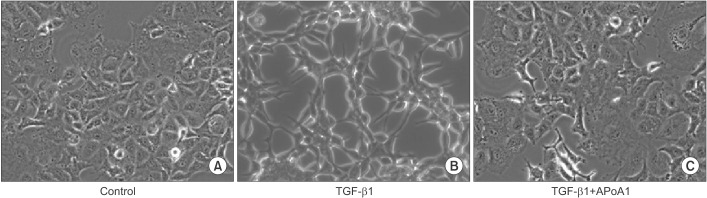
Figure 1 ApoA1 inhibits TGF-β1–induced morphological change in A549 cells. A549 cells were incubated with TGF-β1 (5 ng/mL) with or without ApoA1 (100 ng/mL) for 48 hours. (A) Untreated A549 cells showed a polygonal shape and intact cell-cell adhesion. (B) TGF-β1–treated cells showed a decrease in cell-cell adhesion and a greater spindle shape. (C) Co-treatment of ApoA1-treated cells maintained a polygonal shape and showed an intact cell-cell adhesion similar to the untreated cells (A–C, ×400). ApoA1: apolipoprotein A1; TGF-β1: transforming growth factor β1.
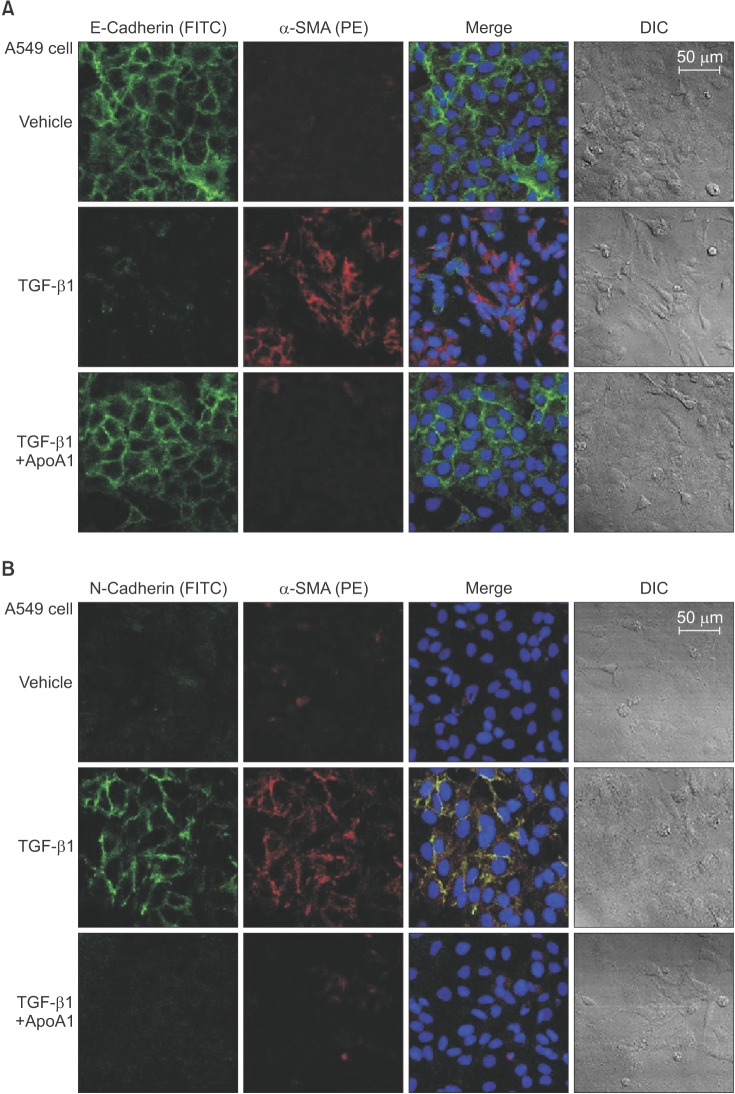
Figure 2 ApoA1 inhibits TGF-β1–induced EMT in A549 cells. A549 cells were treated with TGF-β1 (5 ng/mL) with or without ApoA1 (100 ng/mL) for 48 hours. Expressions of E-cadherin (green) (A), N-cadherin (green) (B), and α-SMA (red) were analyzed by immunofluorescence staining. Treatment with TGF-β1 decreased the expression of the epithelial marker, E-cadherin, and increased the expression of mesenchymal markers such as N-cadherin and α-SMA. ApoA1 inhibited TGF-β1–induced changes in EMT markers similar to control levels (A and B, ×400). ApoA1: apolipoprotein A1; TGF-β1: transforming growth factor β1; EMT: epithelial-to-mesenchymal transition; α-SMA: α-smooth muscle actin; DIC: differential interference contrast.
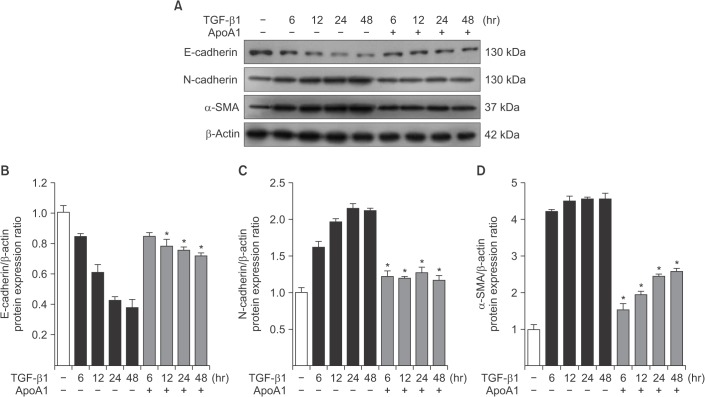
Figure 3 Effects of ApoA1 on EMT-related markers in A549 cells. A549 cells were incubated with TGF-β1 (5 ng/mL) in the absence of serum with or without ApoA1 (100 ng/mL) for up to 48 hours. Stimulation of cells by TGF-β1 down-regulated the epithelial marker, E-cadherin, and up-regulated the mesenchymal markers such as N-cadherin and α-SMA in a time dependent manner. β-Actin was used as a loading control (A). Densitometric analysis of band intensities for E-cadherin (B), N-cadherin (C), and α-SMA band (D). Each bar represents mean±standard error of at least three independent experiments. *p<0.05 versus same time of the TGF-β1–treated group. ApoA1: apolipoprotein A1; EMT: epithelial-to-mesenchymal transition; TGF-β1: transforming growth factor β1; α-SMA: α-smooth muscle actin.
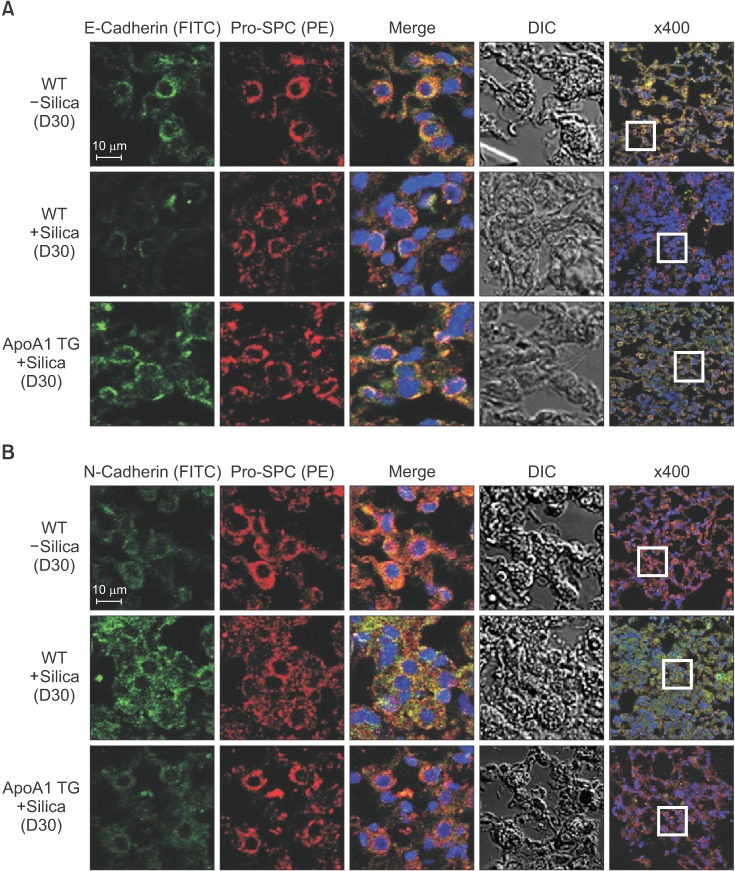
Figure 4 Overexpression of ApoA1 attenuates silica-induced EMT in the mouse lung. Immunofluorescence staining for E-cadherin (green) (A), N-cadherin (green) (B), and pro-SPC (red) in the mouse lung. Treatment with silica decreased the expression of the epithelial marker, E-cadherin, and increased the expression of the mesenchymal markers such as N-cadherin and α-SMA in the WT mouse lung. Overexpression of ApoA1 inhibited TGF-β1–induced changes in EMT markers similar to silica non-treated WT levels (first-fourth columns, ×1,000; fifth columns, ×400). ApoA1: apolipoprotein A1; EMT: epithelial-to-mesenchymal transition; SPC: surfactant protein C; α-SMA: α-smooth muscle actin; WT: wild type; TGF-β1: transforming growth factor β1; DIC: differential interference contrast; TG: transgenic.
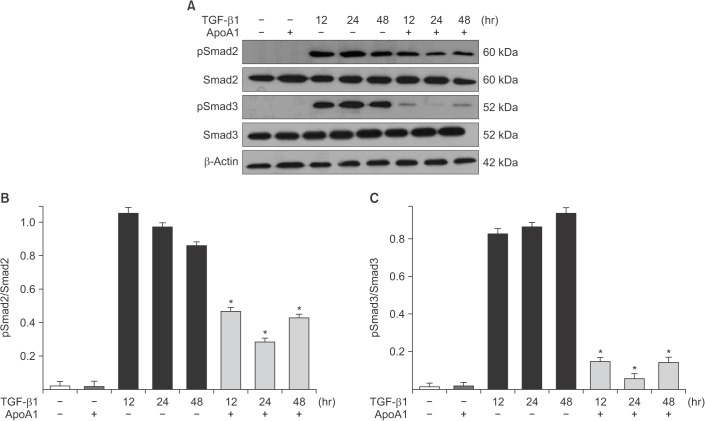
Figure 5 ApoA1 down-regulates the TGF-β1–induced Smad dependent signaling pathway. A549 cells were incubated with TGF-β1 (5 ng/mL) with or without ApoA1 (100 ng/mL) for 48 hours. (A) Phosphorylation of Smad2 and Smad3 occurred after TGF-β1 stimulation and co-treatment with ApoA1 down-regulated the TGF-β1–induced Smad signaling pathway. β-Actin was used as a loading control. Densitometric analysis of band intensities for phosphorylated Smad2 (B) and Smad3 (C). Each bar represents mean±standard error of at least three independent experiments. *p<0.05 versus same time of the TGF-β1–treated group. ApoA1: apolipoprotein A1; TGF-β1: transforming growth factor β1.
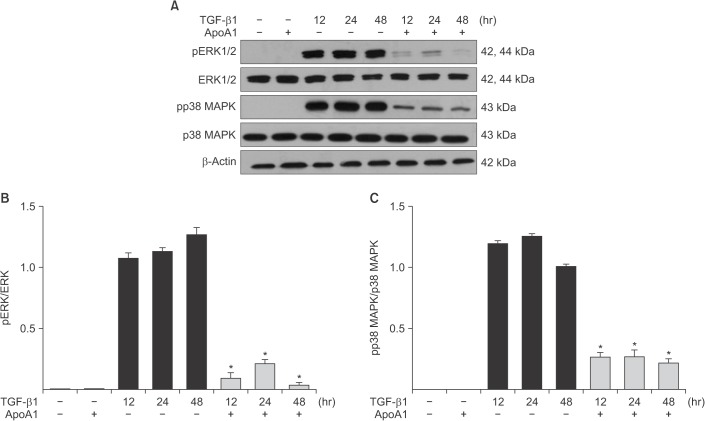
Figure 6 ApoA1 down-regulates the TGF-β1–induced non-Smad signaling pathways. A549 cells were incubated with TGF-β1 (5 ng/mL) with or without ApoA1 (100 ng/mL) for 48 hours. (A) Phosphorylation of ERK1/2 and p38 MAPK occurred after TGF-β1 stimulation and co-treatment with ApoA1 down–regulated the TGF-β1–induced non-Smad signaling pathways. β-Actin was used as a loading control. Densitometric analysis of band intensities for phosphorylated ERK1/2 (B) and p38 MAPK (C). Each bar represents mean±standard error of at least three independent experiments. *p<0.05 versus same time of the TGF-β1–treated group. ApoA1: apolipoprotein A1; TGF-β1: transforming growth factor β1; MAPK: mitogen-activated protein kinase.
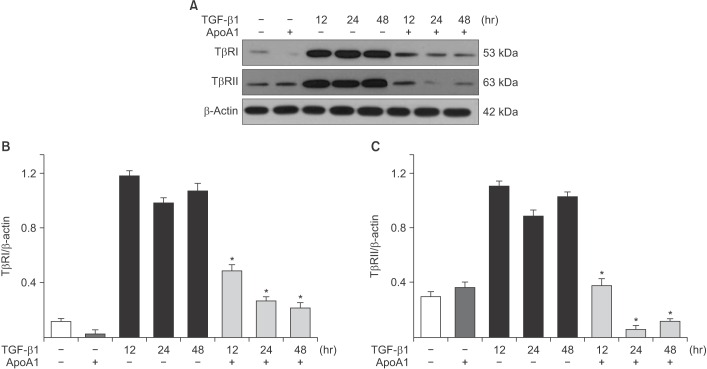
Figure 7 ApoA1 decreases the expression of TβRI and TβRII in A549 cells. A549 cells were incubated with TGF-β1 (5 ng/mL) with or without ApoA1 (100 ng/mL) for 48 hours. (A) TGF-β1 increased both TβRI and TβRII expression and co-treatment with ApoA1 down-regulated TGF-β receptors. β-Actin was used as a loading control. Densitometric analysis of band intensities for TβRI (B) and TβRII (C). Each bar represents mean±SE of at least three independent experiments. *p<0.05 versus same time of the TGF-β1–treated group. ApoA1: apolipoprotein A1; TGF-β1: transforming growth factor β1; TβRI: TGF-β1 receptor type 1; TβRII: TGF-β1 receptor type 2.
Reference
-
1. Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/ fibroblastic cross-talk disorder. Respir Res. 2002; 3:3. PMID: 11806838.
Article2. Harari S, Caminati A. Idiopathic pulmonary fibrosis. Allergy. 2005; 60:421–435. PMID: 15727572.
Article3. Ross R, Everett NB, Tyler R. Wound healing and collagen formation. VI. The origin of the wound fibroblast studied in parabiosis. J Cell Biol. 1970; 44:645–654. PMID: 5415241.4. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med. 1994; 1:71–81. PMID: 8790603.
Article5. Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagenproducing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008; 173:1617–1627. PMID: 19008372.
Article6. Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010; 43:161–172. PMID: 19767450.
Article7. Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004; 15:1–12. PMID: 14694152.
Article8. Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. 2005; 6:56. PMID: 15946381.
Article9. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006; 3:377–382. PMID: 16738204.
Article10. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007; 282:23337–23347. PMID: 17562716.
Article11. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005; 166:1321–1332. PMID: 15855634.12. Khalil N, O'Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, et al. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 1991; 5:155–162. PMID: 1892646.13. Idiopathic Pulmonary Fibrosis Clinical Research Network. Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012; 366:1968–1977. PMID: 22607134.14. Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis: an update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015; 192:e3–e19. PMID: 26177183.
Article15. Kim TH, Lee YH, Kim KH, Lee SH, Cha JY, Shin EK, et al. Role of lung apolipoprotein A-I in idiopathic pulmonary fibrosis: antiinflammatory and antifibrotic effect on experimental lung injury and fibrosis. Am J Respir Crit Care Med. 2010; 182:633–642. PMID: 20463180.16. Lee Eh, Lee EJ, Kim H, Jang A, Koh E, Uh ST, et al. Overexpression of apolipoprotein A1 in the lung abrogates fibrosis in experimental silicosis. PLoS One. 2013; 8:e55827. PMID: 23409054.
Article17. Heldin CH, Moustakas A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012; 347:21–36. PMID: 21643690.18. Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res. 2012; 347:11–20. PMID: 21701805.
Article19. Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009; 21:166–176. PMID: 19237272.20. King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011; 378:1949–1961. PMID: 21719092.
Article21. Spagnolo P, Sverzellati N, Rossi G, Cavazza A, Tzouvelekis A, Crestani B, et al. Idiopathic pulmonary fibrosis: an update. Ann Med. 2015; 47:15–27. PMID: 25613170.
Article22. Noguchi S, Yamauchi Y, Takizawa H. Novel therapeutic strategies for fibrotic lung disease: a review with a focus on epithelial-mesenchymal transition. Recent Pat Inflamm Allergy Drug Discov. 2014; 8:9–18. PMID: 24383438.
Article23. Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014; 69:760–765. PMID: 24334519.
Article24. Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007; 293:L525–L534. PMID: 17631612.25. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003; 425:577–584. PMID: 14534577.26. Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. FASEB J. 2003; 17:1576–1578. PMID: 12824291.27. Lee CM, Park JW, Cho WK, Zhou Y, Han B, Yoon PO, et al. Modifiers of TGF-beta1 effector function as novel therapeutic targets of pulmonary fibrosis. Korean J Intern Med. 2014; 29:281–290. PMID: 24851060.28. Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007; 132:1311–1321. PMID: 17934117.29. Gharaee-Kermani M, Hu B, Phan SH, Gyetko MR. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the myofibroblast. Curr Med Chem. 2009; 16:1400–1417. PMID: 19355895.30. Gomer RH, Lupher ML Jr. Investigational approaches to therapies for idiopathic pulmonary fibrosis. Expert Opin Investig Drugs. 2010; 19:737–745.
Article31. Datta A, Scotton CJ, Chambers RC. Novel therapeutic approaches for pulmonary fibrosis. Br J Pharmacol. 2011; 163:141–172. PMID: 21265830.
Article32. Rojas A, Padidam M, Cress D, Grady WM. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim Biophys Acta. 2009; 1793:1165–1173. PMID: 19339207.33. Sherman CB, Peterson SJ, Frishman WH. Apolipoprotein A-I mimetic peptides: a potential new therapy for the prevention of atherosclerosis. Cardiol Rev. 2010; 18:141–147. PMID: 20395699.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Parthenolide inhibits transforming growth factor β1-induced epithelial-mesenchymal transition in colorectal cancer cells
- Dexamethasone Inhibits TGF-β1-Induced Cell Migration by Regulating the ERK and AKT Pathways in Human Colon Cancer Cells Via CYR61
- Trefoil Factor 1 Suppresses Epithelial-mesenchymal Transition through Inhibition of TGF-beta Signaling in Gastric Cancer Cells
- Inhibitory Effects of Resveratrol on Airway Remodeling by Transforming Growth Factor-β/Smad Signaling Pathway in Chronic Asthma Model
- Aspirin-Triggered Resolvin D1 Inhibits TGF-β1-Induced EndMT through Increasing the Expression of Smad7 and Is Closely Related to Oxidative Stress
