J Clin Neurol.
2013 Jan;9(1):21-25. 10.3988/jcn.2013.9.1.21.
Decreased Metabolism in the Cerebral Cortex in Early-Stage Huntington's Disease: A Possible Biomarker of Disease Progression?
- Affiliations
-
- 1Department of Neurology, Eulji General Hospital, Eulji University School of Medicine, Deajeon, Korea.
- 2Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea.
- 3Department of Nuclear Medicine, Ajou University School of Medicine, Suwon, Korea.
- 4Department of Nuclear Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- 5Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- 6Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. jinwhan.cho@samsung.com
- KMID: 2287566
- DOI: http://doi.org/10.3988/jcn.2013.9.1.21
Abstract
- BACKGROUND AND PURPOSE
Huntington's disease (HD) is an autosomal-dominant inherited neurodegenerative disorder. Genetic analysis of abnormal CAG expansion in the IT15 gene allows disease confirmation even in the preclinical stage. However, because there is no treatment to cure or delay the progression of this disease, monitoring of biological markers that predict progression is warranted.
METHODS
FDG-PET was applied to 13 patients with genetically confirmed HD in the early stage of the disease. We recorded the initial and follow-up statuses of patients using the Independence Scale (IS) of the Unified Huntington's Disease Rating Scale. The progression rate (PR) was calculated as the annual change in the IS. The patients were divided into two groups with faster and slower progression, using the median value of the PR as the cut-off. FDG-PET data were analyzed using regions of interest, and compared among the two patient groups and 11 age- and sex-matched controls.
RESULTS
The mean CAG repeat size in patients was 44.7. The CAG repeat length was inversely correlated with the age at onset as reported previously, but was not correlated with the clinical PR. Compared with normal controls, hypometabolism was observed even at very early stages of the disease in the bilateral frontal, temporal, and parietal cortices on FDG-PET. The decreases in metabolism in the bilateral frontal, parietal, and right temporal cortices were much greater in the faster-progression group than in the slower-progression group.
CONCLUSIONS
A decrease in cortical glucose metabolism is suggested as a predictor for identifying a more rapid form of progression in patients with early-stage HD.
MeSH Terms
Figure
-
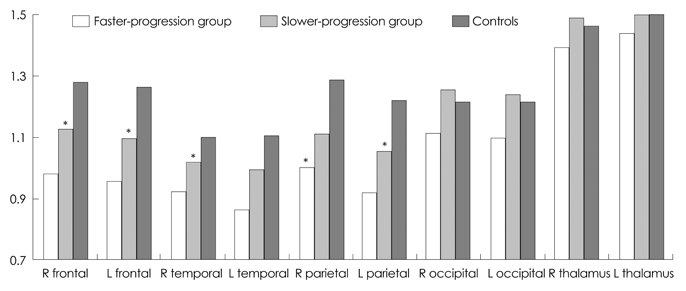
Fig. 1 Comparison of the FDG-PET regional cerebral/cerebellar region-of-interest ratio among faster- and slower-progression Huntington's disease (HD) groups and age- and sex-matched controls. The decreases in metabolism in the bilateral frontal, parietal cortices, and right temporal cortex were significantly greater in the faster-progression group than in the slower-progression group. The metabolism in the occipital cortices and thalami did not differ between the two HD groups. Data are not shown for the bilateral caudate nuclei because of severe decreases in both patient groups. *A statistically significant difference among the groups (p<0.05).
Reference
-
1. The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group. ACMG/ASHG statement. Laboratory guidelines for Huntington disease genetic testing. Am J Hum Genet. 1998. 62:1243–1247.2. Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, et al. A worldwide study of the Huntington's disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994. 330:1401–1406.
Article3. Hersch S, Rosas H, Ferrante R. Neuropathology and pathophysiology of Huntington's disease. 2004. New York: McGraw-Hill.4. Hayden MR, Hewitt J, Stoessl AJ, Clark C, Ammann W, Martin WR. The combined use of positron emission tomography and DNA polymorphisms for preclinical detection of Huntington's disease. Neurology. 1987. 37:1441–1447.
Article5. Mazziotta JC, Phelps ME, Pahl JJ, Huang SC, Baxter LR, Riege WH, et al. Reduced cerebral glucose metabolism in asymptomatic subjects at risk for Huntington's disease. N Engl J Med. 1987. 316:357–362.
Article6. Grafton ST, Mazziotta JC, Pahl JJ, St George-Hyslop P, Haines JL, Gusella J, et al. A comparison of neurological, metabolic, structural, and genetic evaluations in persons at risk for Huntington's disease. Ann Neurol. 1990. 28:614–621.
Article7. Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, et al. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington's disease. Brain. 1996. 119:2085–2095.
Article8. Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, et al. Cerebral cortex and the clinical expression of Huntington's disease: complexity and heterogeneity. Brain. 2008. 131:1057–1068.
Article9. Rosas HD, Liu AK, Hersch S, Glessner M, Ferrante RJ, Salat DH, et al. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002. 58:695–701.
Article10. Martin WR, Clark C, Ammann W, Stoessl AJ, Shtybel W, Hayden MR. Cortical glucose metabolism in Huntington's disease. Neurology. 1992. 42:223–229.
Article11. Kuwert T, Lange HW, Langen KJ, Herzog H, Aulich A, Feinendegen LE. Cortical and subcortical glucose consumption measured by PET in patients with Huntington's disease. Brain. 1990. 113:1405–1423.
Article12. Ashizawa T, Wong LJ, Richards CS, Caskey CT, Jankovic J. CAG repeat size and clinical presentation in Huntington's disease. Neurology. 1994. 44:1137–1143.
Article13. Kieburtz K, MacDonald M, Shih C, Feigin A, Steinberg K, Bordwell K, et al. Trinucleotide repeat length and progression of illness in Huntington's disease. J Med Genet. 1994. 31:872–874.
Article14. Esmaeilzadeh M, Ciarmiello A, Squitieri F. Seeking brain biomarkers for preventive therapy in Huntington disease. CNS Neurosci Ther. 2011. 17:368–386.
Article15. Squitieri F, Cannella M, Frati L. Molecular medicine: predicting and preventing Huntington's disease. Neurol Sci. 2008. 29:205–207.
Article16. Aylward EH, Codori AM, Barta PE, Pearlson GD, Harris GJ, Brandt J. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Arch Neurol. 1996. 53:1293–1296.
Article17. Aylward EH, Brandt J, Codori AM, Mangus RS, Barta PE, Harris GJ. Reduced basal ganglia volume associated with the gene for Huntington's disease in asymptomatic at-risk persons. Neurology. 1994. 44:823–828.
Article18. Antonini A, Leenders KL, Eidelberg D. [11C]raclopride-PET studies of the Huntington's disease rate of progression: relevance of the trinucleotide repeat length. Ann Neurol. 1998. 43:253–255.
Article19. Thieben MJ, Duggins AJ, Good CD, Gomes L, Mahant N, Richards F, et al. The distribution of structural neuropathology in pre-clinical Huntington's disease. Brain. 2002. 125:1815–1828.
Article20. Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003. 60:1615–1620.
Article21. Ciarmiello A, Cannella M, Lastoria S, Simonelli M, Frati L, Rubinsztein DC, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington's disease. J Nucl Med. 2006. 47:215–222.
