Bleeding Tendency of a Light Chain (AL) Amyloidosis Patient Accompanied by Asymptomatic Plasma Cell Myeloma
- Affiliations
-
- 1Department of Laboratory Medicine, Seoul National University College of Medicine, Seoul, Korea. lukekhk@snu.ac.kr
- 2Department of Internal Medicine, Seoul National University College of Medicine, Seoul, Korea.
- KMID: 2053555
- DOI: http://doi.org/10.3343/lmo.2013.3.3.183
Abstract
- We present a case of abrupt-onset hemorrhagic tendency in a patient with amyloidosis who also had asymptomatic plasma cell myeloma. The patient was a 66-yr-old man with no previous history of hemorrhagic tendency and no family history of hemorrhagic disease. On examination, the prothrombin time and activated partial thromboplastin time were found to be prolonged and were not corrected even after a mixing test; moreover, the levels of coagulation factors I, II, V, VII, and X were almost normal. We therefore considered the presence of a nonspecific coagulation inhibitor. Although the von Willebrand factor (vWF) activity and vWF antigen level were normal due to sampling following transfusion, the increased closure time on PFA-100 (Siemens) analysis and the absence of ristocetin-induced platelet aggregation suggested the presence of acquired von Willebrand syndrome (vWS). After chemotherapy, the patient showed alleviation in the bleeding symptoms. Therefore, testing for acquired vWS should be considered when a patient has a history of recent bleeding with underlying amyloidosis.
MeSH Terms
Figure
-
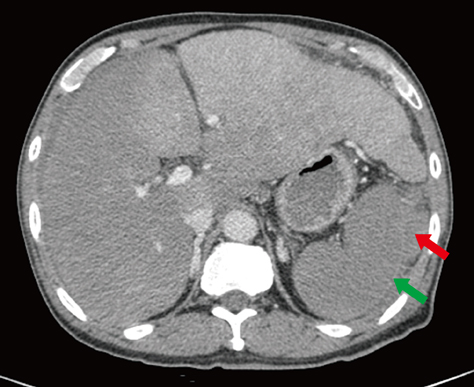
Fig. 1 Abdominal computed tomography showed splenic subcapsular hematoma (red arrow). Spleen showed increase in size, lobulating contour, decreased perfusion with delayed capsular enhancement (green arrow) on portal phase and increased amount of perisplenic fluid.
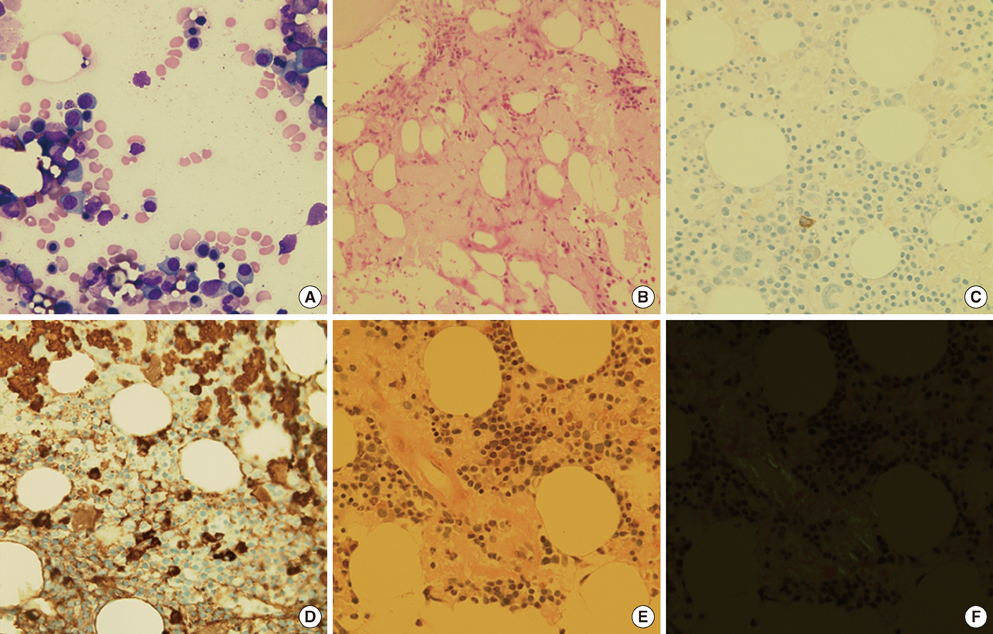
Fig. 2 (A) Bone marrow aspiration showed increased number of plasma cells (21.8% of total nucleated cells, Wright Giemsa, ×400). (B) Bone marrow biopsy showed interstitial deposits of pink amorphous waxy-appearing substances (H&E, ×200). (C, D) Increased clonal plasma cells are negative for cytoplasmic kappa light chain (C) and positive for lambda light chain (D) in bone marrow biopsy (Immunohistochemical stain, ×400). (E, F) Congo red stain produced amyloid pink to red (E) by standard light microscopy and apple-green birefringence (F) under polarized light microscopy (Congo Red, ×400).
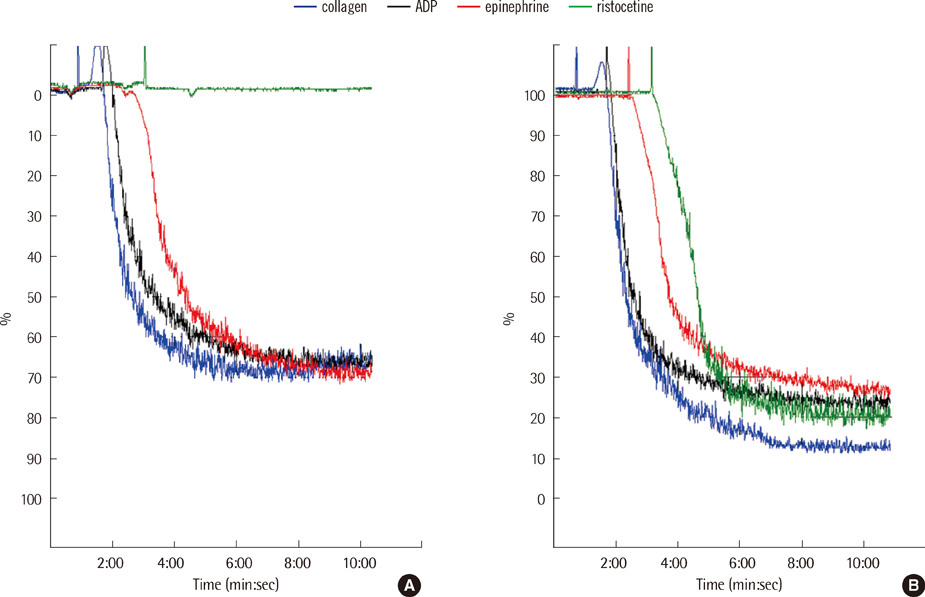
Fig. 3 Results of platelet aggregation test in the patient (A) and a healthy control (B). The patient showed normal aggregation response to 10 µM ADP (71%), 10 µM epinephrine (75%) and 2 µg/mL collagen (72%), but no aggregation response to 1.25 mg/mL ristocetin.
Reference
-
1. Al Dieri R, Peyvandi F, Santagostino E, Giansily M, Mannucci PM, Schved JF, et al. The thrombogram in rare inherited coagulation disorders: its relation to clinical bleeding. Thromb Haemost. 2002; 88:576–582.
Article2. Khoory MS, Nesheim ME, Bowie EJ, Mann KG. Circulating heparan sulfate proteoglycan anticoagulant from a patient with a plasma cell disorder. J Clin Invest. 1980; 65:666–674.
Article3. Hwang KK, Grossman JM, Visvanathan S, Chukwuocha RU, Woods VL Jr, Le DT, et al. Identification of anti-thrombin antibodies in the antiphospholipid syndrome that interfere with the inactivation of thrombin by antithrombin. J Immunol. 2001; 167:7192–7198.
Article4. Sucker C, Hetzel GR, Grabensee B, Stockschlaeder M, Scharf RE. Amyloidosis and bleeding: pathophysiology, diagnosis, and therapy. Am J Kidney Dis. 2006; 47:947–955.
Article5. Enjeti AK, Walsh M, Seldon M. Spontaneous major bleeding in acquired factor X deficiency secondary to AL-amyloidosis. Haemophilia. 2005; 11:535–538.
Article6. Kos CA, Ward JE, Malek K, Sanchorawala V, Wright DG, O'Hara C, et al. Association of acquired von Willebrand syndrome with AL amyloidosis. Am J Hematol. 2007; 82:363–367.
Article7. Gamba G, Montani N, Anesi E, Palladini G, Capezzera M, Soldavini E, et al. Clotting alterations in primary systemic amyloidosis. Haematologica. 2000; 85:289–292.8. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995; 32:45–59.9. Mumford AD, O'Donnell J, Gillmore JD, Manning RA, Hawkins PN, Laffan M. Bleeding symptoms and coagulation abnormalities in 337 patients with AL-amyloidosis. Br J Haematol. 2000; 110:454–460.
Article10. Choufani EB, Sanchorawala V, Ernst T, Quillen K, Skinner M, Wright DG, et al. Acquired factor X deficiency in patients with amyloid light-chain amyloidosis: incidence, bleeding manifestations, and response to high-dose chemotherapy. Blood. 2001; 97:1885–1887.
Article11. Kratzer MA. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function: a rebuttal. J Thromb Haemost. 2006; 4:1845–1846.
Article12. Hayward CP, Harrison P, Cattaneo M, Ortel TL, Rao AK. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J Thromb Haemost. 2006; 4:312–319.
Article13. Favaloro EJ. The utility of the PFA-100 in the identification of von Willebrand disease: a concise review. Semin Thromb Hemost. 2006; 32:537–545.
Article14. Harrison P. The role of PFA-100 testing in the investigation and management of haemostatic defects in children and adults. Br J Haematol. 2005; 130:3–10.
Article15. Federici AB, Rand JH, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost. 2000; 84:345–349.
Article16. Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008; 14:171–232.
Article17. Siaka C, Rugeri L, Caron C, Goudemand J. A new ELISA assay for diagnosis of acquired von Willebrand syndrome. Haemophilia. 2003; 9:303–308.
Article18. Tatewaki W, Takahashi H, Shibata A. Multimeric composition of plasma von Willebrand factor in chronic myeloproliferative disorders. Clin Lab Haematol. 1988; 10:417–425.
Article19. Tefferi A, Hanson CA, Kurtin PJ, Katzmann JA, Dalton RJ, Nichols WL. Acquired von Willebrand's disease due to aberrant expression of platelet glycoprotein Ib by marginal zone lymphoma cells. Br J Haematol. 1997; 96:850–853.
Article20. Scrobohaci ML, Daniel MT, Levy Y, Marolleau JP, Brouet JC. Expression of GpIb on plasma cells in a patient with monoclonal IgG and acquired von Willebrand disease. Br J Haematol. 1993; 84:471–475.
Article21. Pakk HW, Kim IH, Park SH, Lee DS, Park SY, Cho HI, et al. Acquired von Willebrand syndrome associated with amyloidosis. Korean J Hematol. 2009; 44:310–314.
Article22. Federici AB. Acquired von Willebrand syndrome: is it an extremely rare disorder or do we see only the tip of the iceberg. J Thromb Haemost. 2008; 6:565–568.
Article23. Kumar S, Pruthi RK, Nichols WL. Acquired von Willebrand's syndrome: a single institution experience. Am J Hematol. 2003; 72:243–247.
Article24. Hong S, Lee J, Chi H, Lee C, Nah S, Kim Y, et al. Systemic lupus erythematosus complicated by acquired von Willebrand's syndrome. Lupus. 2008; 17:846–848.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- AL amyloidosis: advances in diagnosis and management
- A Case of Multiple Myeloma of Kappa Light Chain Type Associated with Gastric Amyloidosis and Acute Renal Failure and Pathologic Fracture Due to Femur Plasmacytoma
- Oral Phase Dysphagia, the First Manifestation of Systemic Amyloidosis Associated with Multiple Myeloma
- A Case of Macroglossia due to Amyloidosis Associated with Multiple Myeloma
- A Case of Gastrointestinal Amyloidosis in Asymptomatic Multiple Myeloma
