The Blocking of c-Met Signaling Induces Apoptosis through the Increase of p53 Protein in Lung Cancer
- Affiliations
-
- 1Brain Korea 21 Project for Biomedical Science, Korea University College of Medicine, Seoul, Korea. yhk0215@korea.ac.kr
- 2Genomic Research Center for Lung and Breast/Ovarian Cancers, Korea University College of Medicine, Seoul, Korea.
- 3Laboratory of Radiation Tumor Physiology, Korea Institute of Radiological and Medical Science, Korea University College of Medicine, Seoul, Korea.
- 4Division of Oncology/Hematology, Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea.
Abstract
- PURPOSE
c-Met is an attractive potential target for novel therapeutic inhibition of human cancer, and c-Met tyrosine kinase inhibitors (TKIs) are effective growth inhibitors of various malignancies. However, their mechanisms in anticancer effects are not clear. In the present study, we investigated the possibility that blocking c-Met signaling induces p53-mediated growth inhibition in lung cancer.
MATERIALS AND METHODS
The growth inhibitory effects of c-Met TKI (SU11274) on lung cancer cells and a xenograft model were assessed using the MTT assay, flow cytometry, and terminal deoxyribonucleotide transferase-mediated nick-end labeling staining. The role of p53 protein in the sensitivity of c-Met TKI (SU11274) was examined by Western blot analysis and immunohistochemistry.
RESULTS
SU11274 significantly induced apoptosis in A549 cells with wild-type p53, compared with that in Calu-1 cells with null-type p53. SU11274 increased p53 protein by enhancing the stability of p53 protein. Increased p53 protein by SU11274 induced up-regulation of Bax and PUMA expression and down-regulation of Bcl-2 expression, subsequently activating caspase 3. In p53 knock-out and knock-in systems, we confirmed that SU11274 caused apoptosis through the p53-mediated apoptotic pathway. Likewise, in the A549 xenograft model, SU11274 effectively shrank tumor volume and induced apoptosis via increased p53 protein expression. Blocking c-Met signaling increased the level of p53 protein.
CONCLUSION
Our finding suggested that p53 plays an important role in SU11274-induced apoptosis, and p53 status seems to be related to the sensitivity to SU11274 in lung cancer.
Keyword
MeSH Terms
-
Apoptosis
Blotting, Western
Caspase 3
Down-Regulation
Flow Cytometry
Growth Inhibitors
Humans
Indoles
Lung
Lung Neoplasms
Molecular Targeted Therapy
Piperazines
Protein-Tyrosine Kinases
Puma
Sulfonamides
Transplantation, Heterologous
Tumor Burden
Tumor Suppressor Protein p53
Up-Regulation
Caspase 3
Growth Inhibitors
Indoles
Piperazines
Protein-Tyrosine Kinases
Sulfonamides
Tumor Suppressor Protein p53
Figure
-
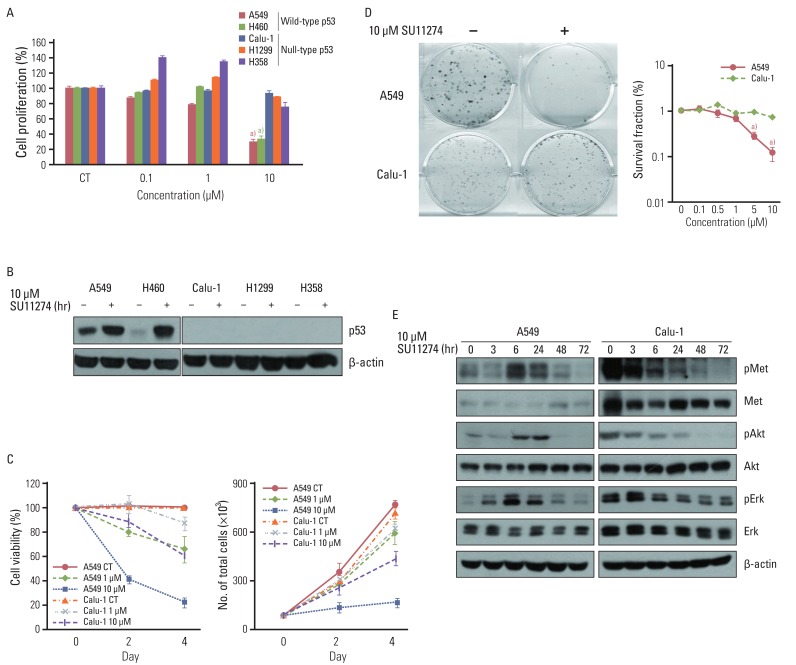
Fig. 1 Effect of SU11274 on growth inhibition in lung cancer cells. (A) The result of MTT assay in lung cancer cells with different p53 statuses treated with the indicated concentrations of SU11274 for 72 hr. Columns, mean value of six identical wells of a single representative experiment; Bars, standard error. a)p<0.001 for comparisons between SU11274-treated and untreated cells. (B) The expression of p53 protein in lung cancer cells. Cells were treated with 10 µM SU11274 for 24 hr, and cell extracts were immunoblotted using p53 antibody. β-actin serves as a loading control. (C) Results of cell counting in A549 and Calu-1 cells treated with 1 and 10 µM SU11274 for 48 hr and 96 hr. Points, mean value of three experiments; bar, standard error, this data presented p < 0.05; CT, control. (D) Results of colony-forming assay in A549 and Calu-1 cells treated with indicated concentrations of SU11274 for ~3 weeks. The surviving fraction (SF) was calculated from the following formula: SF=number of colonies formed/number of cells seeded×plating efficiency of control group. Points, mean values of three experiments; bar, standard error. a)p<0.001. (E) A549 and Calu-1 cells were treated with 10 µM SU11274 for indicated time. Whole cell lysates were prepared and separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and immunoblotted for MET, Akt, Erk and their phosphorylated forms. β-actin was used as a loading control.
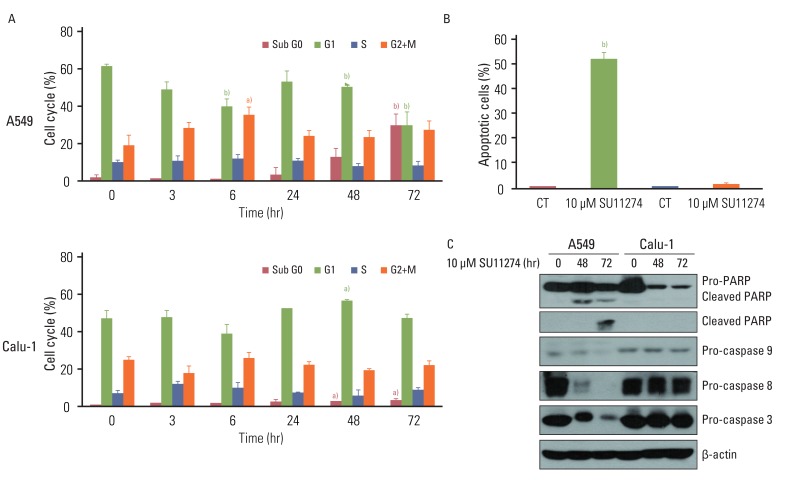
Fig. 2 Effects of SU11274 on apoptosis in A549 and Calu-1 cells. (A) Results of cell cycle analysis using propidium iodide staining in SU11274-treated A549 and Calu-1 cells. Cells were treated with 10 µM SU11274 for indicated period of time, and both adherent and floating cells were harvested for analysis of cell cycle distribution. Columns, mean value of three experiments; bar, standard error. a)p<0.05, b)p<0.001. (B) The results of apoptosis in A549 and Calu-1 cells assessed using APO-BRDU kit. A549 and Calu-1 cells were treated with 10 µM SU11274 for 72 hr, and both adherent and floating cells were harvested. Columns, mean value of three identical experiments; bars, standard error; CT, control. b)p<0.001 for comparisons between SU11274-treated and untreated cells. (C) Expression of poly(ADP-ribose) polymerase (PARP), caspase 9, caspase 8, and caspase 3 in A549 and Calu-1 cells. Cells were treated with 10 µM SU11274 for 48 and 72 hr. Both adherent and floating cells were harvested.
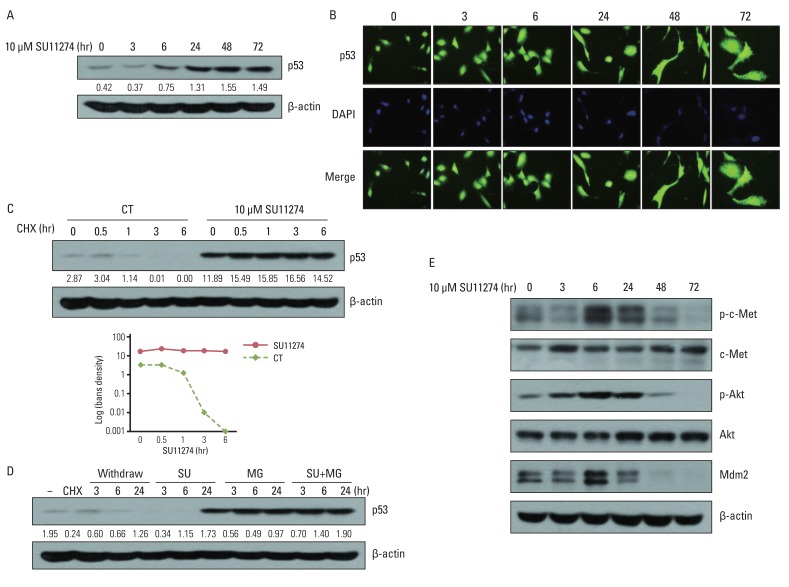
Fig. 3 Effects of SU11274 on p53 protein in A549 cells. (A) The expression of p53 protein in SU11274-treated A549 cells. Cells were treated with SU11274 for the indicated periods of time. Whole cell lysates were prepared and separated by 10% sodium dodecyl sulfatepolyacrylamide gel electrophoresis, and immunoblotted using p53 antibodies. β-actin serves as a loading control. Bands were quantified by densitometry. (B) The effects of SU11274 on cellular localization of p53 protein in A549 cells. SU11274-treated A549 cells immunostained with anti-p53 antibody (green fluorescence) and DAPI (blue fluorescence) in a time-dependent manner, and analyzed by fluorescence microscopy to determine the cellular localization of p53. (C) p53 stability in SU11274-treated A549 cells. Cells were treated with 10 µM SU11274 for 24 hr, followed by 10 µg/mL cycloheximide (CHX) for the indicated periods of time. CT, control. (D) p53 increase in SU11274-treated A549 cells. Cells were treated with 10 µg/mL CHX for 6 hr, followed by change of new growing medium only and new growing medium containing 10 µM SU11274, 10 µM MG132, or both for indicated times. Each band was quantified by densitometry. SU, SU11274; MG, MG132. (E) The expression of phospho-c-Met (p-c-Met), c-Met, phospho-Akt (p-Akt), Akt, and mdm2 protein. Cells were treated with 10 µM SU11274 for the indicated time.
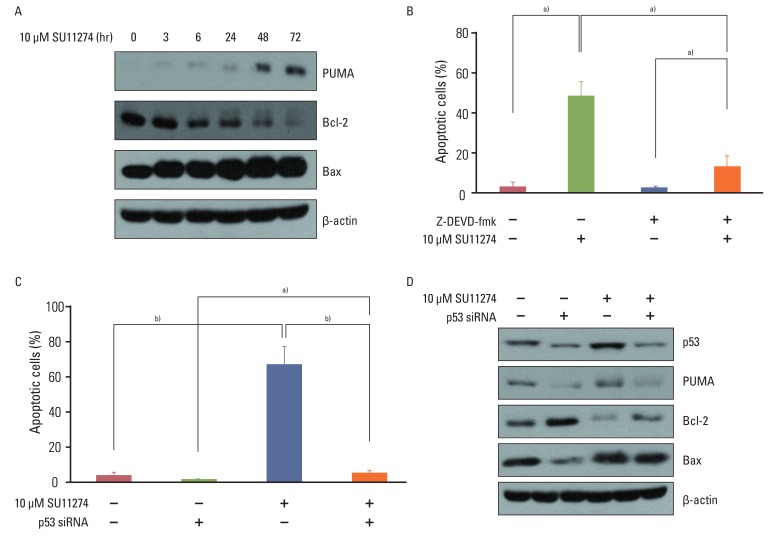
Fig. 4 The role of p53 in SU11274-mediated apoptosis in A549 cells. (A) Expression of Bax, PUMA, and Bcl-2 in SU11274-treated A549 cells. Cells were treated with 10 µM SU11274 for the indicated times. (B) Effects of caspase 3 on apoptosis in SU11274-treated A549 cells. Cells were treated with 10 µM SU11274, 2 µM Z-DEVD-fmk (caspase 3 inhibitor), or both for 72 hr. Both adherent and floating cells were harvested and evaluated by the APO-BRDU kit. Columns, mean value of three experiments; bar, standard error. a)p<0.05. (C) Effects of p53 siRNA on apoptosis in SU11274-induced apoptosis using APO-BRDU kit. Cells were transiently transfected with scramble siRNA or p53 siRNA. After 24 hr, cells were treated with 10 µM SU11274 for 72 hr. Both adherent and non-adherent cells were harvested. Columns, mean value of three experiments; bar, standard error. a)p<0.05, b)p<0.005. (D) Expression of p53, Bax, PUMA, and Bcl-2 protein in p53 siRNA-transfected A549 cells after SU11274 treatment.
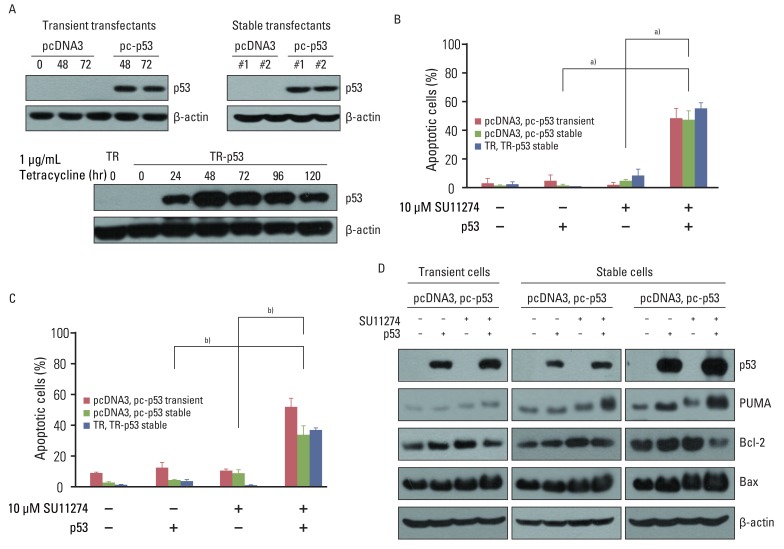
Fig. 5 Effects of exogenous p53 protein on SU11274-mediated apoptosis. (A) Expression of p53 protein in p53 transfectants. Cells were transfected with pcDNA3, pcDNA3-p53 (pc-p53) expression vectors, pcDNA6/TR (TR), or pcDNA6/TR-pcDNA4/TO-p53 (TR-p53) expression vectors. To prepare transient transfectants, cells were transfected with pcDNA3 and pc-p53. After 48 and 72 hr, cells were harvested and immunoblotted using p53 antibodies. Stable transfectants, described in Materials and Methods, were harvested and analyzed by immunoblotting. One µg/mL Tet analogue tetracycline was applied to TR and TR-p53 stable transfectants for 24 hr and the cells were harvested at the indicated times. Cell extracts were analyzed by immunoblotting using p53 antibody. β-actin was used as a loading control. (B) Effects of exogenous p53 protein on SU11274-induced apoptosis in empty vector and p53 transfectants. TR and TR-p53 stable transfectants were pretreated with 1 µg Tet analogue tetracycline for 24 hr and total cells were treated with 10 µM SU11274 for 72 hr. Both adherent and non-adherent cells were harvested. Columns, mean value of three experiments; bar, standard error. a)p<0.005. (C) Result of cell cycle analysis using propidium iodide staining in p53 transfected Calu-1 cell after SU11274 treatment. Cells were transiently transfected with pcDNA3 and pc-p53 DNA. After 24 hr, cells were treated with 10 µM SU11274 for 72 hr. Stable p53 transfectants were also treated with 10 µM SU11274 for 72 hr. Both adherent and floating cells were harvested for analysis of cell cycle distribution. Columns, mean value of three experiments; bar, standard error. b)p<0.001. (D) Expression of p53, Bax, PUMA, and Bcl-2 proteins in empty vector and p53 transfectants. Cells were treated with 10 µM SU11274 for 72 hr.
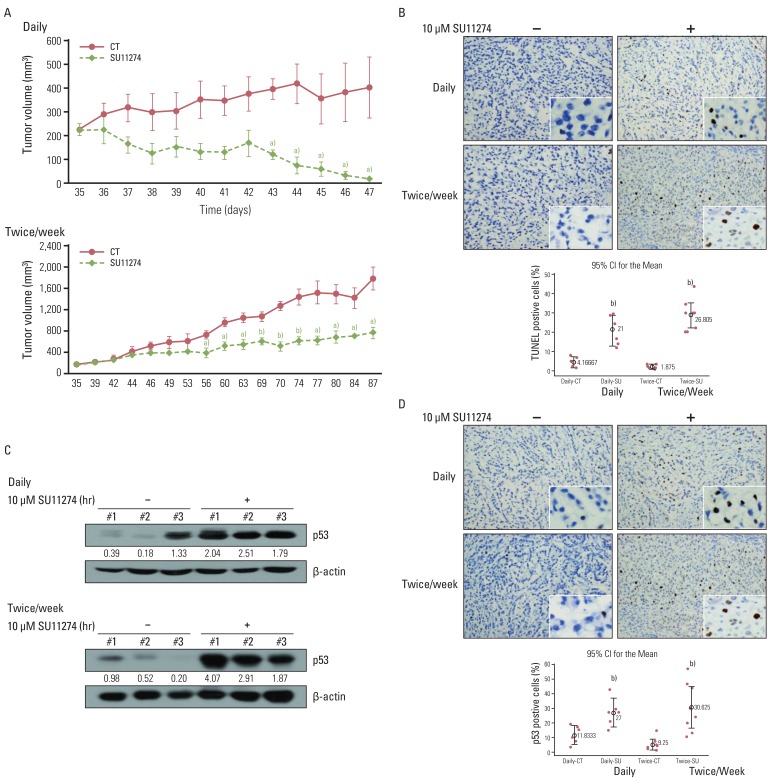
Fig. 6 Effects of SU11274 on tumor growth in A549 xenograft model. (A) Change of tumor volume after treatment with SU11274 daily and twice weekly for the indicated periods in the xenograft model. SU11274 (100 µg per xenograft) was administered on two schedules through intratumoral injections; it was administered daily for 13 days or twice weekly for 53 days. The tumor volume was recorded at the indicated times. n=7 (dimethyl sulfoxide [DMSO]-treated group [n=3] and SU11274-treated group [n=4]). Animal experiments were performed done twice independently. Points, mean value of three or four identical mice; bars, standard error. a)p<0.05, b)p<0.001. (B) Tumors were harvested on the last day, and apoptosis was assessed using the terminal deoxyribonucleotide transferase-mediated nick-end labeling (TUNEL) assay. Cells stained brown indicate apoptotic cells. CT, control; SU, SU11274. (C) Expression of p53 in tumor tissue harvested from A549 xenograft. Tumor tissue was lysed with homogenizer, resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and immunoblotted using p53 antibodies. Bands were quantified by densitometry. (D) Harvested tumor tissue was immunohistochemically examined with anti-p53 antibody. Brown color indicates the expression of p53 protein. The percentage of positive cells was determined from the following formula: The percentage of terminal deoxyribonucleotide transferase-mediated nick-end labeling/p53-positive cells=positive staining cells/total cells×100. Box, interquartile range of six-eight identical mice;⌖, mean value of six or eight identical mice. b)p<0.001 for comparisons between SU11274-treated and untreated mice.
Reference
-
1. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991; 251:802–804. PMID: 1846706.2. Furge KA, Zhang YW, Vande Woude GF. Met receptor tyrosine kinase: enhanced signaling through adapter proteins. Oncogene. 2000; 19:5582–5589. PMID: 11114738.
Article3. Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005; 225:1–26. PMID: 15922853.
Article4. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039–1043. PMID: 17463250.5. Eder JP, Vande Woude GF, Boerner SA, LoRusso PM. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009; 15:2207–2214. PMID: 19318488.
Article6. Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R. Downstream signalling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: implications for tumour invasion. Br J Cancer. 2007; 97:368–377. PMID: 17667909.
Article7. Puri N, Khramtsov A, Ahmed S, Nallasura V, Hetzel JT, Jagadeeswaran R, et al. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007; 67:3529–3534. PMID: 17440059.
Article8. Trotman LC, Pandolfi PP. PTEN and p53: who will get the upper hand? Cancer Cell. 2003; 3:97–99. PMID: 12620402.
Article9. Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003; 3:117–130. PMID: 12620407.
Article10. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001; 98:11598–11603. PMID: 11504915.
Article11. Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993; 7:1126–1132. PMID: 8319905.
Article12. Moumen A, Patane S, Porras A, Dono R, Maina F. Met acts on Mdm2 via mTOR to signal cell survival during development. Development. 2007; 134:1443–1451. PMID: 17329361.
Article13. Rong S, Donehower LA, Hansen MF, Strong L, Tainsky M, Jeffers M, et al. Met proto-oncogene product is overexpressed in tumors of p53-deficient mice and tumors of Li-Fraumeni patients. Cancer Res. 1995; 55:1963–1970. PMID: 7728766.14. Peruzzi B, Bottaro DP. Targeting the c-Met signaling pathway in cancer. Clin Cancer Res. 2006; 12:3657–3660. PMID: 16778093.
Article15. Ma PC, Maulik G, Christensen J, Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003; 22:309–325. PMID: 12884908.16. Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003; 63:7345–7355. PMID: 14612533.17. Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, et al. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007; 67:2081–2088. PMID: 17332337.
Article18. Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001; 13:332–337. PMID: 11343904.
Article19. Momand J, Wu HH, Dasgupta G. MDM2: master regulator of the p53 tumor suppressor protein. Gene. 2000; 242:15–29. PMID: 10721693.20. Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene. 1993; 8:307–318. PMID: 8426740.21. Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, El Dinali M, et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009; 69:3021–3031. PMID: 19318576.
Article22. Zimmer Y, Vaseva AV, Medova M, Streit B, Blank-Liss W, Greiner RH, et al. Differential inhibition sensitivities of MET mutants to the small molecule inhibitor SU11274. Cancer Lett. 2010; 289:228–236. PMID: 19783361.
Article23. Xiong L, Kou F, Yang Y, Wu J. A novel role for IGF-1R in p53-mediated apoptosis through translational modulation of the p53-Mdm2 feedback loop. J Cell Biol. 2007; 178:995–1007. PMID: 17846171.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Quercetin induces cell death by caspase-dependent and p38 MAPK pathway in EGFR mutant lung cancer cells
- The Immunohistochemical analysis for the expression of survivin, an inhibitor of apoptosis protein, in non-small cell lung cancer
- p53 signaling is involved in leptin-induced growth of hepatic and breast cancer cells
- The Role of p53 in Marijuana Smoke Condensates-induced Genotoxicity and Apoptosis
- The Antitumor Effect of C-terminus of Hsp70-Interacting Protein via Degradation of c-Met in Small Cell Lung Cancer
