Impact of Enzyme Replacement Therapy on Linear Growth in Korean Patients with Mucopolysaccharidosis Type II (Hunter Syndrome)
- Affiliations
-
- 1Department of Pediatrics, Hanyang University Guri Hopistal, Hanyang University College of Medicine, Guri, Korea.
- 2Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. jindk@skku.edu
- 3Department of Pediatrics, Kwandong University College of Medicine, Myongji Hospital, Goyang, Korea.
- 4Department of Medical Genetics, Ajou University Hospital, Ajou University School of Medicine, Suwon, Korea.
- 5Department of Pediatrics, Kwandong University College of Medicine, Cheil General Hospital & Woman's Health care Center, Seoul, Korea.
- KMID: 1789984
- DOI: http://doi.org/10.3346/jkms.2014.29.2.254
Abstract
- Hunter syndrome (or mucopolysaccharidosis type II [MPS II]) arises because of a deficiency in the lysosomal enzyme iduronate-2-sulfatase. Short stature is a prominent and consistent feature in MPS II. Enzyme replacement therapy (ERT) with idursulfase (Elaprase(R)) or idursulfase beta (Hunterase(R)) have been developed for these patients. The effect of ERT on the growth of Korean patients with Hunter syndrome was evaluated at a single center. This study comprised 32 patients, who had received ERT for at least 2 yr; they were divided into three groups according to their ages at the start of ERT: group 1 (<6 yr, n=14), group 2 (6-10 yr, n=11), and group 3 (10-20 yr, n=7). The patients showed marked growth retardation as they got older. ERT may have less effect on the growth of patients with the severe form of Hunter syndrome. The height z-scores in groups 2 and 3 revealed a significant change (the estimated slopes before and after the treatment were -0.047 and -0.007, respectively: difference in the slope, 0.04; P<0.001). Growth in response to ERT could be an important treatment outcome or an endpoint for future studies.
Keyword
MeSH Terms
-
Adolescent
Body Height
Child
Child, Preschool
Demography
Enzyme Replacement Therapy
Humans
Iduronate Sulfatase/*therapeutic use
Infant
Male
Mild Cognitive Impairment/etiology
Mucopolysaccharidosis II/complications/diagnosis/*therapy
Mutation
Phenotype
Protein Isoforms/therapeutic use
Republic of Korea
Young Adult
Iduronate Sulfatase
Protein Isoforms
Figure
-
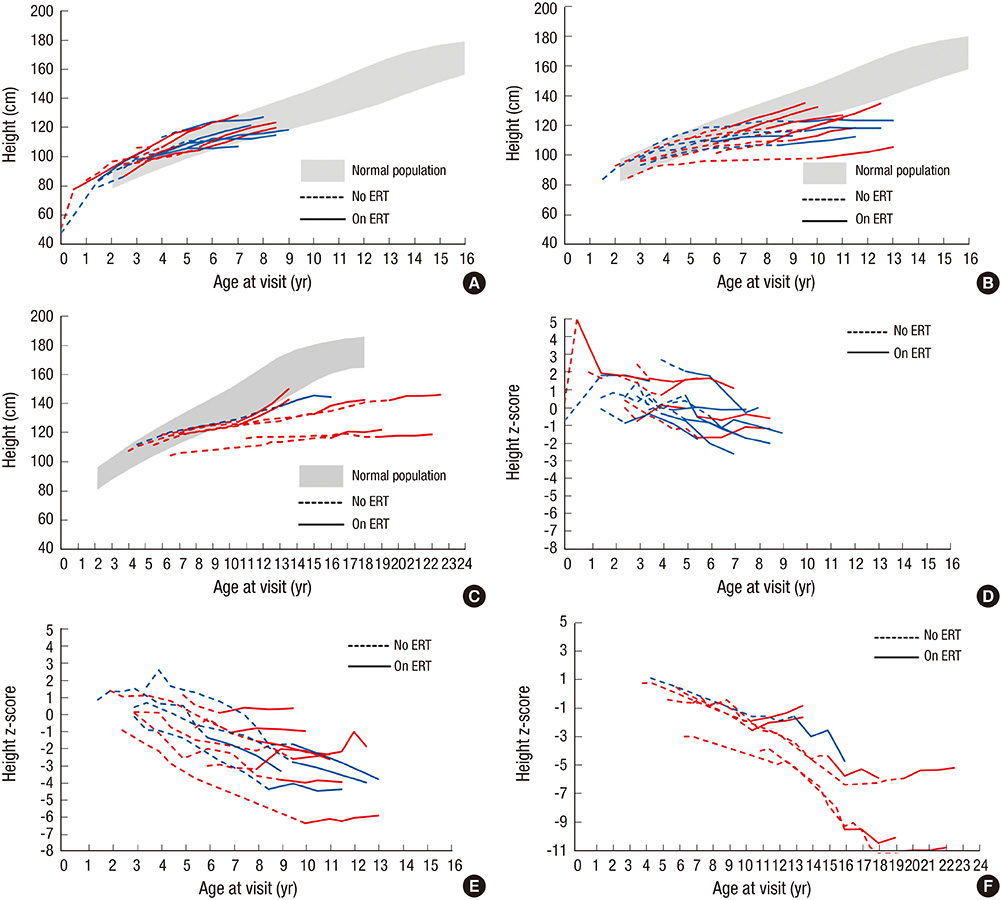
Fig. 1 Growth charts and z-scores for height for Hunter syndrome patients. (A) Growth charts for group 1 (aged less than 6 yr at initiation of ERT). (B) Growth charts for group 2 (aged 6-10 yr at initiation of ERT). (C) Growth charts for group 3 (aged 10-20 yr at initiation of ERT). (D) Z scores for group 1 (aged less than 6 yr at initiation of ERT). (E) Z scores for group 2 (aged 6-10 yr at initiation of ERT). (F) Z scores for group 3 (aged 10-20 yr at initiation of ERT). The dotted lines indicate patient growth before enzyme replacement therapy (ERT), and the continuous lines denote the growth while receiving ERT. The shaded area represents the 3rd to the 97th percentiles of height in boys based on normative data from Korean references. The red lines represent the patients with the attenuated form, and the blue lines denote those with the severe form.
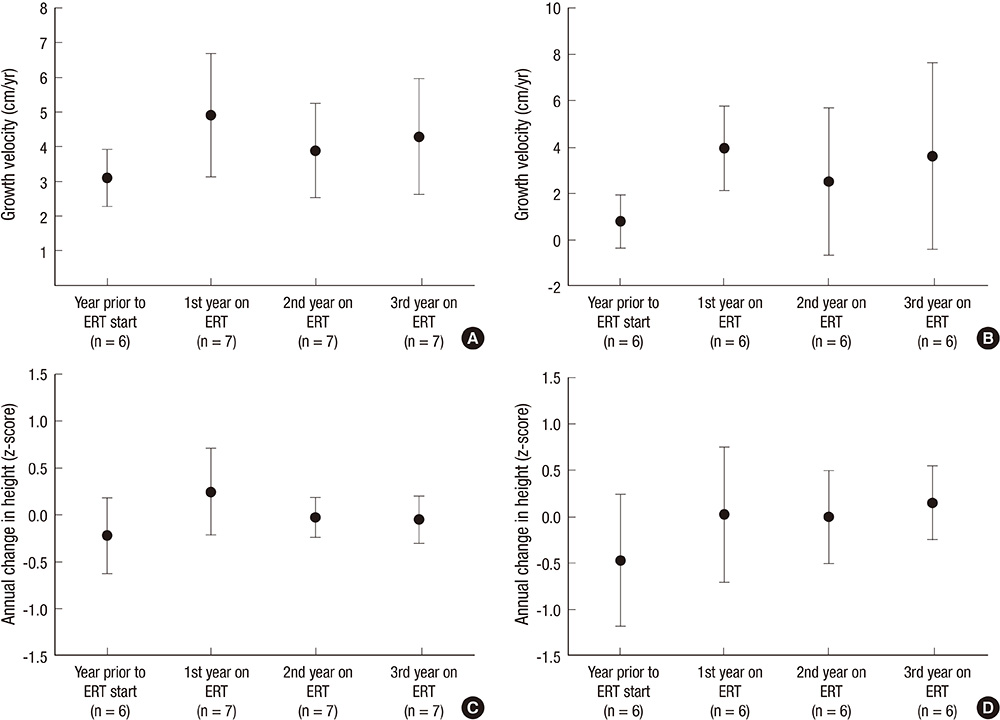
Fig. 2 Yearly growth velocity and z-score of the patients with the attenuated form of groups 2 and 3. (A) Yearly growth velocity for group 2 (aged 6-10 yr at initiation of ERT). (B) Yearly growth velocity for group 3 (aged 10-20 yr at initiation of ERT). (C). Z scores for group 2 (aged 6-10 yr at initiation of ERT). (D) Z scores for group 3 (aged 10-20 yr at initiation of ERT).
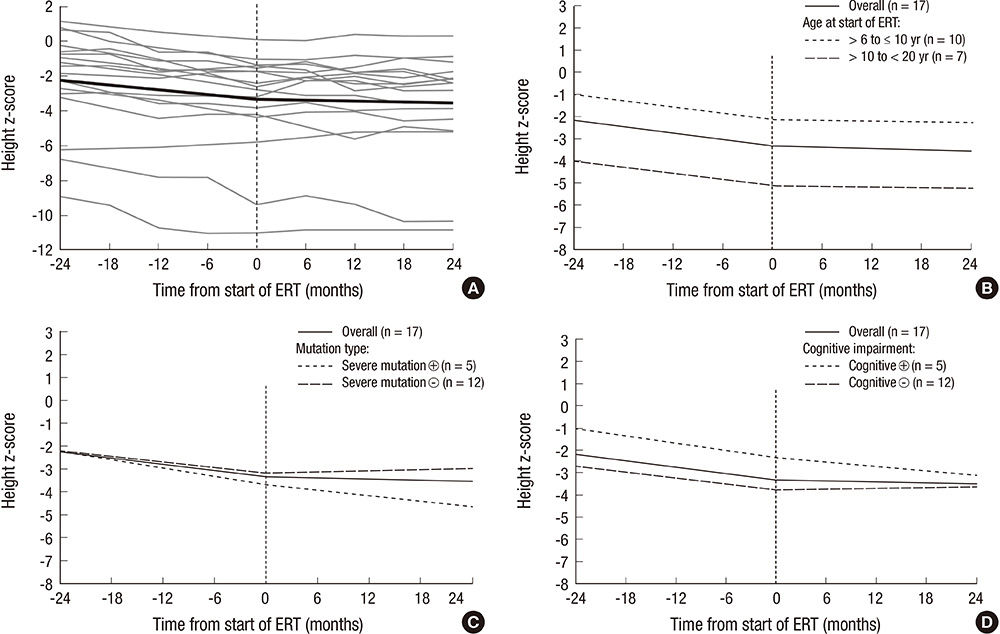
Fig. 3 Regression plot showing the height z-scores before and after initiation of enzyme replacement therapy (ERT) in 17 patients aged 6-20 yr at treatment start. (A) The gray lines show the regression plot for each patient. The slope of the regression was significantly changed after the ERT compared with before the treatment (difference in z-score, 0.04; P < 0.001). (B) Impact of age at start of ERT. (C) Impact of type of mutation. (D) Impact of cognitive impairment.
Reference
-
1. Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999; 105:151–156.2. Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, Kohlschütter A, Kampmann C, Beck M. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005; 28:1011–1017.3. Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ, Chao MC, Chiu PC, Lin SJ, Tsai LP, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet A. 2009; 149A:960–964.4. Neufeld E, Muenzer J. The metabolic and molecular bases of inherited disease. In : Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The mucopolysaccharidoses. New York: McGraw-Hill;2001. p. 3421–3452.5. Schwartz IV, Ribeiro MG, Mota JG, Toralles MB, Correia P, Horovitz D, Santos ES, Monlleo IL, Fett-Conte AC, Sobrinho RP, et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl. 2007; 96:63–70.6. Schulze-Frenking G, Jones SA, Roberts J, Beck M, Wraith JE. Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis. 2011; 34:203–208.7. Rozdzynska A, Tylki-Szymanska A, Jurecka A, Cieslik J. Growth pattern and growth prediction of body height in children with mucopolysaccharidosis type II. Acta Paediatr. 2011; 100:456–460.8. Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. HOS Investigators. Initial report from the Hunter Outcome Survey. Genet Med. 2008; 10:508–516.9. Gordon M, Crouthamel C, Post EM, Richman RA. Psychosocial aspects of constitutional short stature: social competence, behavior problems, self-esteem, and family functioning. J Pediatr. 1982; 101:477–480.10. Stabler B, Clopper RR, Siegel PT, Stoppani C, Compton PG, Underwood LE. Academic achievement and psychological adjustment in short children: the National Cooperative Growth Study. J Dev Behav Pediatr. 1994; 15:1–6.11. Stephen MD, Varni JW, Limbers CA, Yafi M, Heptulla RA, Renukuntla VS, Bell CS, Brosnan PG. Health-related quality of life and cognitive functioning in pediatric short stature: comparison of growth-hormone-naïve, growth-hormone-treated, and healthy samples. Eur J Pediatr. 2011; 170:351–358.12. Sohn YB, Cho SY, Park SW, Kim SJ, Ko AR, Kwon EK, Han SJ, Jin DK. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter syndrome). Orphanet J Rare Dis. 2013; 8:42.13. Muenzer J, Wraith JE, Beck M, Giugliani R, Harmatz P, Eng CM, Vellodi A, Martin R, Ramaswami U, Gucsavas-Calikoglu M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006; 8:465–473.14. Muenzer J, Beck M, Eng CM, Giugliani R, Harmatz P, Martin R, Ramaswami U, Vellodi A, Wraith JE, Cleary M, et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet Med. 2011; 13:95–101.15. Jones SA, Parini R, Harmatz P, Giugliani R, Fang J, Mendelsohn NJ. HOS Natural History Working Group on behalf of HOS Investigators. The effect of idursulfase on growth in patients with Hunter syndrome: data from the Hunter Outcome Survey (HOS). Mol Genet Metab. 2013; 109:41–48.16. Sohn YB, Ki CS, Kim CH, Ko AR, Yook YJ, Lee SJ, Kim SJ, Park SW, Yeau S, Kwon EK, et al. Identification of 11 novel mutations in 49 Korean patients with mucopolysaccharidosis type II. Clin Genet. 2012; 81:185–190.17. Kim J, Park MR, Kim DS, Lee JO, Maeng SH, Cho SY, Han Y, Ahn K, Jin DK. IgE-mediated anaphylaxis and allergic reactions to idursulfase in patients with Hunter syndrome. Allergy. 2013; 68:796–802.18. Lee OJ, Kim SJ, Sohn YB, Park HD, Lee SY, Kim CH, Ko AR, Yook YJ, Lee SJ, Park SW, et al. A study of the relationship between clinical phenotypes and plasma iduronate-2-sulfatase enzyme activities in Hunter syndrome patients. Korean J Pediatr. 2012; 55:88–92.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A case of Hunter's Syndrome: Mucopolysaccharidosis type II
- A case of Hunter syndrome
- Cochlear Implantation via the Transmeatal Approach in an Adolescent with Hunter Syndrome—Type II Mucopolysaccharidosis
- Clinical and Laboratory Features of Korean Mucopolysaccharidoses (MPSs)
- A Case of Hunter's Syndrome
