J Korean Med Sci.
2007 Dec;22(6):946-951. 10.3346/jkms.2007.22.6.946.
The Genotype and Clinical Phenotype of Korean Patients with Familial Hypokalemic Periodic Paralysis
- Affiliations
-
- 1Department of Pediatrics, College of Medicine, The Catholic University of Korea, Seoul, Korea. byungcl@catholic.ac.kr
- 2Department of Neurology, Seoul National University, Seoul, Korea.
- 3Division of Nephrology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- KMID: 1785751
- DOI: http://doi.org/10.3346/jkms.2007.22.6.946
Abstract
- Familial hypokalemic periodic paralysis (HOPP) is a rare autosomal-dominant disease characterized by reversible attacks of muscle weakness occurring with episodic hypokalemia. Mutations in the skeletal muscle calcium (CACNA1S) and sodium channel (SCN4A) genes have been reported to be responsible for familial HOPP. Fifty-one HOPP patients from 20 Korean families were studied to determine the relative frequency of the known mutations and to specify the clinical features associated with the identified mutations. DNA analysis identified known mutations in 12 families: 9 (75%) were linked to the CACNA1S gene and 3 (25%) to the SCN4A gene. The Arg528His mutation in the CACNA1S gene was found to be predominant in these 12 families. Additionally, we have detected one novel silent exonic mutation (1950C>T) in the SCN4A gene. As for a SCN4A Arg669His mutation, incomplete penetrance in a woman was observed. Characteristic clinical features were observed both in patients with and without mutations. This study presents comprehensive data on the genotype and phenotype of Korean families with HOPP.
MeSH Terms
Figure
-
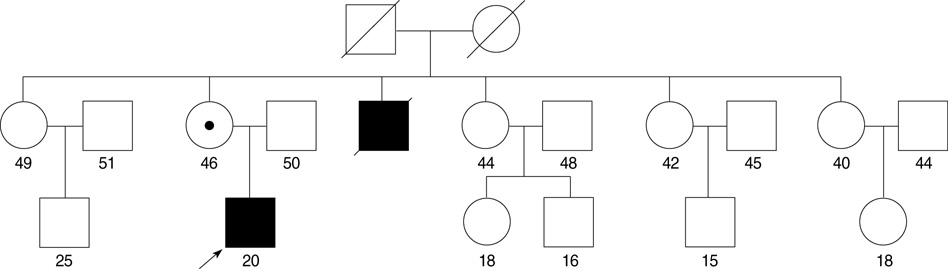
Fig. 1 Pedigree of a family with the SCN4A Arg669His mutation. Dark symbols represent the affected individuals. The symbol with a dot designates an asymptomatic carrier. Slash marks represent deceased individuals. The proband is indicated by an arrow. The age of the family members is designated by the number.
Reference
-
1. Lapie P, Lory P, Fontaine B. Hypokalemic periodic paralysis: an autosomal dominant muscle disorder caused by mutations in a voltage-gated calcium channel. Neuromuscul Disord. 1997. 7:234–240.
Article2. Jen J, Ptacek L. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Metabolic and molecular bases of inherited disease. 2001. 8th ed. New York: McGraw-Hill;5223–5238.3. Talbott JH. Periodic paralysis. Medicine. 1941. 20:85–96.
Article4. Sternberg D, Maisonobe T, Jurkat-Rott K, Nicole S, Launay E, Chauveau D, Tabti N, Lehmann-Horn F, Hainque B, Fontaine B. Hypokalemic periodic paralysis type2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. 2001. 124:1091–1099.5. Fontaine B, Vale-Santos JM, Jurkat-Rott K, Reboul J, Plassart E, Rime CS, Heine R, Guimaraes J, Weissenbach J, Baumann N, Fardeau M, Lehmann-Horn F. Mapping of hypokalemic periodic paralysis (HypoPP) locus to chromosome 1q31-q32 in three European families. Nat Genet. 1994. 6:267–272.6. Ptacek LJ, Tawil R, Griggs RC, Engel AG, Layzer RB, Kwiecinski H, McManis PG, Santiago L, Moore M, Fouad G, Bradley P, Leppert MF. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. 1994. 77:863–868.
Article7. Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogen K, Powers PA, Lapie P, Vale-Santos JE, Weissenbach J, Fontaine B. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. 1994. 3:1415–1419.
Article8. Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, Ebers GC. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. 1999. 53:1932–1936.
Article9. Jurkat-Rott K, Mitrovic N, Hang C, Kouzmekine A, Iaizzo P, Herzog J, Lerche H, Nicole S, Vale-Santos J, Chauveau D, Fontaine B, Lehmann-Horn F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA. 2000. 97:9549–9554.
Article10. Kim MK, Lee SH, Park MS, Kim BC, Cho KH, Lee MC, Kim JH, Kim SM. Mutation screening in Korean hypokalemic periodic paralysis patients: a novel SCN4A Arg672Cys mutation. Neuromuscul Disord. 2004. 14:727–731.
Article11. Miller TM, Dias da Silva MR, Miller HA, Kwiecinski H, Mendell JR, Tawil R, McManis P, Griggs RC, Angelini C, Servidei S, Petajan J, Dalakas MC, Ranum LP, Fu YH, Ptacek LJ. Correlating phenotype and genotype in the periodic paralyses. Neurology. 2004. 63:1647–1655.
Article12. Grosson CL, Esteban J, Mckenna-Yasek D, Gusella JF, Brown RH Jr. Hypokalemic periodic paralysis mutations: confirmation of mutation and analysis of founder effect. Neuromuscul Disord. 1996. 6:27–31.
Article13. Fouad G, Dalakas M, Servidei S, Mendell JR, Van den Bergh P, Angelini C, Alderson K, Griggs RC, Tawil R, Gregg R, Hogan K, Powers PA, Weinberg N, Malonee W, Ptacek LJ. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromusc Disord. 1997. 7:33–38.14. Bendahhou S, Cummins TR, Griggs RC, Fu YH, Ptacek LJ. Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann Neurol. 2001. 50:417–420.
Article15. Elbaz A, Vale-Santos J, Jurkat-Rott K, Lapie P, Ophoff RA, Bady B, Links TP, Piussan C, Vila A, Monnier N. Hypokalemic periodic paralysis and the dihydropyridine receptor (CACNL1A 3): genotype/ phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 caucasian families. Am J Hum Genet. 1995. 56:374–380.16. Cannon SC. An expanding view for the molecular basis of familial periodic paralysis. Neuromuscul Disord. 2002. 12:533–543.
Article17. Ober KP. Thyrotoxic periodic paralysis in the United States: report of 7 cases and review of the literature. Medicine (Baltimore). 1992. 71:109–120.
Article18. Paul B, Hirudayaraj P, Baig MW. Thyrotoxic periodic paralysis: an unusual presentation of weakness. Emerg Med J. 2003. 20:E7.
Article19. Kim JB, Lee KY, Hur JK. A Korean family of hypokalemic periodic paralysis with mutation in a voltage-gated calcium channel (R1239G). J Korean Med Sci. 2005. 20:162–165.
Article20. Kim SH, Kim UK, Chae JJ, Kim DJ, Oh HY, Kim BJ, Lee CC. Identification of mutations including de novo mutations in Korean patients with hypokalemic periodic paralysis. Nephrol Dial Transplant. 2001. 16:939–944.21. Venance SL, Jurkat-Rott K, Lehmann-Horn F, Tawil R. SCN4-Aassociated hypokalemic periodic paralysis merits a trial of acetazolamide. Neurology. 2004. 63:1977.
Article22. Pagani F, Stuani C, Tzetis M, Kanavakis E, Efthymiadou A, Doudounakis S, Casals T, Baralle FE. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum Mol Genet. 2003. 12:1111–1120.
Article23. D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999. 96:5598–5603.24. Poskanzer DC, Kerr DN. Periodic paralysis with response to spironolactone. Lancet. 1961. 2:511–513.
Article25. Torres CF, Griggs RC, Moxley RT, Bender AN. Hypokalemic periodic paralysis exacerbated by acetazolamide. Neurology. 1981. 31:1423–1428.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Hypokalemic Familial Periodic Paralysis: A Report of 4 members in a family
- An atypical phenotype of hypokalemic periodic paralysis caused by a mutation in the sodium channel gene SCN4A
- Two Cases of Familial Hypokalemic Periodic Paralysis
- Hypokalemic Periodic Paralysis Developed in a Patient with Neurogenic Diabetes Insipidus
- Genetics of Channelopathy: Familial Periodic Paralysis
