J Korean Med Sci.
2005 Feb;20(1):162-165. 10.3346/jkms.2005.20.1.162.
A Korean Family of Hypokalemic Periodic Paralysis with Mutation in a Voltage-gated Calcium Channel (R1239G)
- Affiliations
-
- 1Department of Pediatrics, College of Medicine, The Catholic University of Korea, Seoul, Korea. jkhur@catholic.ac.kr
- KMID: 1781747
- DOI: http://doi.org/10.3346/jkms.2005.20.1.162
Abstract
- Hypokalemic periodic paralysis (HOPP) is a rare disease characterized by reversible attacks of muscle weakness accompanied by episodic hypokalemia. Recent molecular work has revealed that the majority of familial HOPP is due to mutations in a skeletal muscle voltage-dependent calcium-channel: the dihydropyridine receptor. We report a 13-yr old boy with HOPP from a family in which 6 members are affected in three generations. Genetic examination identified a nucleotide 3705 C to G mutation in exon 30 of the calcium channel gene, CACNA1S. This mutation predicts a codon change from arginine to glycine at the amino acid position #1239 (R1239G). Among the three known mutations of the CACNA1S gene, the R1239G mutation was rarely reported. This boy and the other family members who did not respond to acetazolamide, showed a marked improvement of the paralytic symptoms after spironolactone treatment.
MeSH Terms
-
Acetazolamide/pharmacology
Adolescent
Arginine/chemistry
Calcium Channels/chemistry/*genetics
Codon
Exons
Family Health
Female
Glycine/chemistry
Humans
Hypokalemia/metabolism
Hypokalemic Periodic Paralysis/*diagnosis/*genetics
Korea
Male
Muscle, Skeletal/metabolism
Mutation
Pedigree
Protein Structure, Tertiary
Sequence Analysis, DNA
Spironolactone/pharmacology
Figure
-
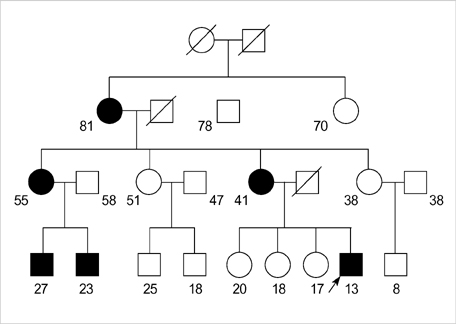
Fig. 1 Pedigree of the HOPP family. The dark symbols are affected individuals. The proband is indicated by an arrow. The age of the family members is designated by the number.
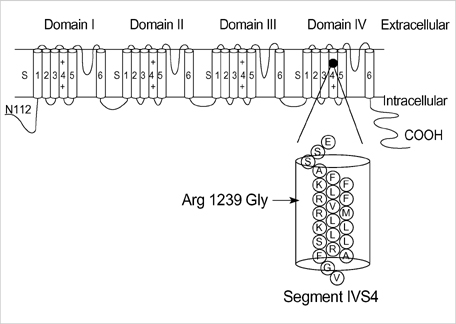
Fig. 2 Schematic diagram of the DHP receptor α1-subunit composed of four domains of an internal homology (DI to DIV) connected by intracellular loops. The R1239G mutation substitutes a positively-charged arginine located within segment DIVS4 by a neutral glycine.
Cited by 1 articles
-
The Genotype and Clinical Phenotype of Korean Patients with Familial Hypokalemic Periodic Paralysis
June-Bum Kim, Man-Ho Kim, Soon Ju Lee, Dae-Joong Kim, Byung Churl Lee
J Korean Med Sci. 2007;22(6):946-951. doi: 10.3346/jkms.2007.22.6.946.
Reference
-
1. Talb H. Periodic paralysis. Medicine. 1941. 20:85–96.
Article2. Jen J, Ptacek L. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Channelopathies. Metabolic and molecular bases of inherited disease. 2001. 8th ed. New York: McGraw-Hill;5223–5238.3. Lapie P, Lory P, Fontaine B. Hypokalemic periodic paralysis: an autosomal dominant muscle disorder caused by mutations in a voltage-gated calcium channel. Neuromusc Disord. 1997. 7:234–240.
Article4. Sternberg D, Maisonobe T, Jurkat-Rott K, Nicole S, Launay E, Chauveau D, Tabti N, Lehmann-Horn F, Hainque B, Fontaine B. Hypokalemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. 2001. 124:1091–1099.5. Caciotti A, Morrone A, Domenici R, Donati MA, Zammarchi E. Severe prognosis in a large family with hypokalemic periodic paralysis. Muscle Nerve. 2003. 27:165–169.
Article6. Bulman DE, Scoggan KA, van Oene MD, Nicolle MW, Hahn AF, Tollar LL, Ebers GC. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. 1999. 53:1932–1936.
Article7. Jurkat-Rott K, Lehmann-Horn F, Elbaz A, Heine R, Gregg RG, Hogan K, Powers PA, Lapie P, Vale-Santos JE, Weissenbach J, Fontaine B. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. 1994. 3:1415–1419.
Article8. Fontaine B, Vale Santos JM, Jurkat-Rott K, Reboul J, Plassart E, Rime CS, Elbaz A, Heine R, Guimaraes J, Weissenbach J, Baumann N, Fardeau M, Lehmann-Horn F. Mapping of the hypokalemic periodic paralysis to chromosome 1q31-q32 in three European families. Nat Genet. 1994. 6:267–272.9. Ptacek LJ, Tawil R, Griggs RC, Engel AG, Layzer RB, Kwiecinski H, McManis PG, Santiago L, Moore M, Fouad G, Bradley P, Leppert MF. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. 1994. 77:863–868.
Article10. Elbaz A, Vale-Santos J, Jurkat-Rott K, Lapie P, Ophoff RA, Bady B, Links TP, Piussan C, Vila A, Monnier N. Hypokalemic periodic paralysis and the dihydropyridine receptor (CACNL1A 3): genotype/phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 caucasian families. Am J Hum Genet. 1995. 56:374–380.11. Grosson CS, Esteban J, Mckenna-Yasek D, Gusella JF, Brown RH. Hypokalemic periodic paralysis mutations: confirmation of mutation and analysis of founder effect. Neuromusc Disord. 1996. 6:27–31.
Article12. Fouad G, Dalakas M, Servidei S, Mendell JR, Van den Bergh P, Angelini C, Alderson K, Griggs RC, Tawil R, Gregg R, Hogan K, Powers PA, Weinberg N, Malonee W, Ptacek LJ. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromusc Disord. 1997. 7:33–38.13. Bendahhou S, Cummins TR, Hahn AF, Langlois S, Waxman SG, Ptacek LJ. Sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann Neurol. 2001. 50:417–420.
Article14. Seo SK, Lee GH, Jeong HY, Kim SW, Kim KH. A case of familial hypokalemic periodic paralysis. J Korean Pediatr Soc. 1989. 32:1012–1016.15. Kim KK, Lee JH, Lee SS, Roh JK, Lee SB, Myung H. Two cases of familial hypokalemic periodic paralysis. J Korean Neurol Sci. 1990. 8:180–184.16. Kim SH, Kim UK, Chae JJ, Kim DJ, Oh HY, Kim BJ, Lee CC. Identification of mutations including de novo mutations in Korean patients with hypokalemic periodic paralysis. Nephrol Dial Transplant. 2001. 16:939–944.17. Catteral WA. Structure and function of voltage-sensitive ion channels. Science. 1988. 242:50–61.
Article18. Perez-Reyes E, Kim HS, Lacerda AE, Horne W, Wei XY, Rampe D, Campbell KP, Brown AM, Birnbaumer L. Induction of calcium currents by the expression of the α1-subunit of the dihydropyridine receptor from skeletal muscle. Nature. 1989. 340:233–236.
Article19. Griggs RC, Engel WK, Resnick JS. Acetazolamide treatment of hypokalemic periodic paralysis. Ann Intern Med. 1970. 73:39–48.
Article20. Tricarico D, Barbieri M, Camerino DC. Acetazolamide opens the muscular KCa2+ channel: a novel mechanism of action that may explain the therapeutic effect of the drug in hypokalemic periodic paralysis. Ann Neurol. 2000. 48:304–312.
Article21. Links TP, Smit AJ, Molenaar WM, Zwarts MJ, Oosterhuis HJ. Familial hypokalemic periodic paralysis. Clinical, diagnostic and therapeutic aspects. J Neurol Sci. 1994. 122:33–43.22. Poskanzer DC, Kerr DN. Periodic paralysis with response to spironolactone. Lancet. 1961. 2:511–513.
Article23. Torres CF, Griggs RC, Moxley RT, Bender AN. Hypokalemic periodic paralysis exacerbated by acetazolamide. Neurology. 1981. 31:1423–1428.
Article24. Rischbieth RH. Hypokalemic periodic paralysis unresponsive to acetazolamide. Clin Exp Neurol. 1981. 18:87–90.25. Vern BA, Danon MJ, Hanlon K. Hypokalemic periodic paralysis with unusual responses to acetazolamide and sympathomimetics. J Neurol Sci. 1987. 81:159–172.
Article26. Tawil R, McDermott MP, Brown R Jr, Shapiro BC, Ptacek LJ, McManis PG, Dalakas MC, Spector SA, Mendell JR, Hahn AF, Griggs RC. Randomized trials of dichlorphenamide in the periodic paralysis. Working Group on Periodic Paralysis. Ann Neurol. 2000. 47:46–53.27. Lambert C, Blanloeil Y, Horber RK, Berard L, Reyford H, Pinaud M. Malignant hyperthermia in a patient with hypokalemic periodic paralysis. Anesth Analg. 1994. 79:1012–1014.
Article28. Horton B. Anesthetic experiences in a family with hypokalemic familial periodic paralysis. Anesthesiology. 1977. 47:308–310.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Genetics of Channelopathy: Familial Periodic Paralysis
- An atypical phenotype of hypokalemic periodic paralysis caused by a mutation in the sodium channel gene SCN4A
- An Arg528His Mutation of the CACNL1A3 Gene in a Korean Family with Hypokalemic Periodic Paralysis
- Electrophysiological Changes by Exercise and Cold Provocation Test in a Patient with Hyperkalemic Periodic Paralysis
- Familial hyperkalemic periodic paralysis caused by a de novo mutation in the sodium channel gene SCN4A
